<< Back to MOTIFvations Blog Home Page
Oncohistones: Histone Mutations & Their Oncogenic Effects
By Anne-Sophie Ay-Berthomieu, Ph.D.
June 24, 2021
Share ShareTable of Contents:
-
Oncohistones: Mutated Histones & Their Role in Cancer
-
What Is the Physiological Role of Histone Proteins?
-
What Are the Oncohistones?
-
Histone H3
-
Histone H2A
-
Histone H2B
-
Histone H4
-
What Are the Consequences of Histone Mutations in Cancer Development?
-
Summary: Understanding the Variety of Oncohistone Types as Tools in Drug Discovery & Personalized Medicine
Oncohistones: Mutated Histones & Their Role in Cancer
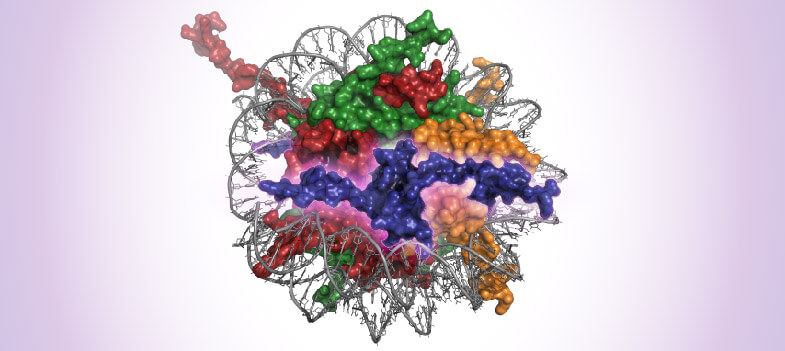
In every cell, genomic DNA is wrapped around a cluster of histone proteins into a basic unit, the nucleosome, which constitutes the structure of chromatin. The nucleosome core is an octamer of histones containing dimers of the histones H2A, H2B, H3, and H4 wrapped with 147bp of DNA. The nucleosomes are stabilized by the linker histone H1. Chromatin is a flexible structure that both protects DNA integrity and regulates transcriptional activity. This flexibility is partly due to the nucleosome, and in particular, histone post-translational modifications (PTMs). Considering its crucial role in basic cellular processes, it is not surprising that chromatin deregulation plays a pivotal role in several human pathologies, including cancer.
Cancer is one of the leading causes of death in the western world. It is characterized by genome instability, transcriptional deregulation, and uncontrolled cell growth. Depending on the cancer type, different gene and amino acid mutations have been identified and studied for diagnostic and prognostic purposes. In B Chronic Lymphocytic Leukemia, immunoglobulin gene mutations are associated with the grade of the disease. In Non-Small Cell Lung Carcinoma, EGFR gene is mutated in 50% of cases in the Asian population and 12% of cases in the Caucasian population, leading to overactivation of the EGF pathway and cell growth acceleration. Interestingly, in pediatric cancers, somatic mutations in histones have been widely described to have a dominant effect on the non-mutated copy of the gene and to be associated with poor prognosis. Histones containing these types of mutations are commonly referred to as “Oncohistones”.
In this article, we will briefly review the biological functions of histones, and we will describe the different types of histone mutations, their role(s) in cancer development and how they can potentially be used as diagnostic and prognostic tools.
What Is the Physiological Role of Histone Proteins?
The histone protein family is comprised of histones H1, H2A, H2B, H3, and H4, representing half of the eucaryotic chromosome’s mass and one of the most evolutionary conserved protein families. Histones are encoded by 73 genes organized in clusters on chromosomes 1, 6, and 12. A total of 16 genes code for H2A, 22 genes for H2B, 14 genes for H3, 15 genes for H4, and 55 genes for H1. Given the importance of histones in cell function, it is not surprising to observe such redundancy in histone coding regions.
The nucleosome core is an octamer of histones comprised of dimers of the histones H2A, H2B, H3, and H4. These histones display a globular domain that is involved in histone-DNA interaction and an N-Terminal tail which is subject to post-translational mutations (PTMs). Histone PTMs are controlled by enzymatic protein modifiers called “writers”, “erasers,” and “readers”. Histones can be acetylated, methylated, phosphorylated, and citrullinated among many other emerging modifications. These PTMs function by regulating chromatin accessibility and therefore transcriptional activity. The nucleosome also maintains genomic integrity, so its destabilization can lead to genome alterations. Histone H1 is a non-nucleosome histone and displays the least conserved sequence across species. H1 maintains DNA wrapping around the nucleosome and stabilizes chromatin structure.
In addition to canonical histones, several histone variants have been identified with significant differences in their sequence. They are incorporated in the nucleosome in a DNA-replication-independent way and replace existing nucleosomal subunits. They can be tissue-specific in their distribution, and unlike canonical histones which are expressed in S-phase, histone variants are expressed throughout the cell cycle. These exchanges of variants in place of other histones are tightly regulated by chaperone proteins and chromatin remodelers. In contrast to canonical histones, these variants are coded by only one gene, containing introns and generated via splicing. The majority of histone variants have been identified for H2A and H3, including 8 variants for H2A and 6 for H3. Two variants were discovered for H2B, 11 for H1, but so far none are known for H4. Like canonical histones, these variants are well conserved across species. Histone variants can have a direct effect on the chromatin structure, stability, remodeling, and can therefore influence transcription regulation.

What Are the Oncohistones?
Oncohistone is a term used when referring to a range of histone alterations implicated in cancer development. These include missense mutations in histone genes or their variants. Cancer-associated point mutations have been found in all 4 core histones, linker histones, and their respective variants. These mutations may affect the activity of reader, writer, and eraser proteins responsible for histone post-translational modifications (PTMs) of histone tails, with significant effects on transcriptional activity and signaling pathways that depend on these modifications. Histone variants themselves may also be inappropriately expressed or incorporated in the chromatin. Beginning in 2012, somatic mutations in histones coding genes have been discovered, with more and more studies underlining the role of these mutations in different kinds of cancer and their role in tumorigenesis. Let’s look at several of the most widely characterized histone mutations.
Histone H3
H3K27M
H3 is the most frequently mutated histone associated with cancers, suggesting a major role for histone H3 in chromatin assembly and transcription regulation. The first H3 mutation on lysine 27 (K27), was discovered in 2012 by two independent labs, St. Jude Children's Research Hospital in Memphis, USA, and McGill University and Genome Quebec Innovation Centre in Montreal, Canada. This mutation substitutes a methionine in place of lysine (K27M) and was found to be associated with pediatric glioblastoma. This mutation was confirmed by many studies and was also found in other cancers, including chondroblastoma, head and neck squamous cell carcinoma, pediatric soft tissue sarcoma, and others. K27M is found in 30% of pediatric high-grade glioblastoma (HGG), mostly in the H3F3A gene coding for the H3.3 variant. It was also detected in 80% of patients with diffuse intrinsic pontine glioma, an aggressive form of HGG, and associated with mutations in TP53, PDGFRA, ACVR1, and BCOR. This mutation doesn’t only affect paediatric tumors but is also implicated in adult cancers such as glioma, acute myeloid leukemia, and melanoma and was associated with a more aggressive phenotype. K27M leads to reduced H3K27 methylation, increase in H3K27ac and DNA hypomethylation, and consequently to gene activation with EZH2 inhibition by a negative-dominant effect.
H3K36M – H3G34
Other mutations were also discovered in the N-Terminal of the histone H3 sequence, the most studied being found at K36 and G34. These induce deregulation in the histone PTM patterns, altering transcription and chromatin accessibility. K36M is detected in 95% of chondroblastoma, and at lower frequency in pediatric soft tissue carcinoma, head and neck squamous cell carcinoma, melanoma, bladder, and colorectal cancer. In chondroblastoma, the mutation is mainly found in the H3F3B gene coding for H3.3 variants, whereas in the other type of cancers, the mutation concerns the H3.1 variant. H3.3K36M has a dominant-negative effect on H3K36me2/3, inhibiting the activity of SETD2 and interfering with H3K27 methylation. G34 mutation is found in children and young adult tumors where it is frequently substitute to R or V. G34R/V was shown to be correlated with ATRX/DAXX mutation in pHGG. Other G34 transitions exist including G34W and G34L, which are found mostly in the H3F3A gene in 90% of giant cell tumors of bone, a tumor type that leads to massive bone destruction. G34W increases the proliferative and migration capacities of these benign tumors and impairs osteogenic differentiation. G34 mutation induces a decrease in K36 methylation by blocking SETD2 binding. Contrary to the above mutations, G34 mutations don’t have a dominant-negative effect but only deregulate K36 methylation locally.
Mutations in the Globular Domain
Some residues in the globular domain of H3 are also found to be mutated at a high rate, including E97, E105, R131, suggesting that not only histone PTM regulation is also important for nucleosome structure. These types of mutations interfere with nucleosome integrity and nucleosome/DNA interactions, leading to chromatin higher structure alteration.
Histone H2A
H2A mutations are less common than mutations in H3 yet they are found in bladder cancer, endometrial carcinomas, and in head and neck cancers. E121 in the C-Terminal tail of H2A transitions to Q, K, and D. H2A is the only histone which bears both an N-Terminal and C-Terminal tail. The C-Terminal tail is known to participate in higher-order chromatin structure, suggesting that E121 mutations could have a role in the destabilization of this structure. Another frequent mutation is R29Q. This arginine is methylated by PRMT6 to repress transcription, whereas the transition to Q prevents this repression.The globular domain of H2A is also affected by mutations at K74 and K75. These lysines are substituted by asparagine and can alter H2A-H3 and H2A-DNA interaction. In uterine and ovarian carcinomas, R4H, K16T, and E57Q have been identified. R4 and K16 are amino acids targeted by post-translational modification. The substitution should alter this process. E57 is involved in structural integrity maintenance. The H2AZ.1 histone variant also shows a high rate of R80C mutation that leads to unstable nucleosomes. Until now, no functional studies showed the role of these mutations in tumorigenesis. However, frequencies of these mutations in cancer suggest an oncogenic role may yet be identified.
Histone H2B
Unlike histone H3, which displays mutations both in the globular domain and tail, H2B mutations are mainly located in the globular regions. E76, E113, F70, and F71 are the most frequently found in cancer. E76, E70, and E71 are located at the interface of H2B-H4, and mutation of these residues induces a distortion of the interface, leading to nucleosome instability. E113 mutation targets the acidic patch at the surface of the H2A/H2B dimer, leading to nucleosome remodeling, DNA damage repair, and chromatin condensation deregulation.The only H2B PTM residue found so far to be mutated in cancer is E2, which is an ADP-ribosylation site. However, no functional studies have been performed as yet. G53 is also frequently mutated, in particular in pancreatic ductal cancer, where G53D is found in 4.5% of patients. At a physiological level, this mutation increases the migration capabilities of the cell. At a molecular level, it enhances transcription activity by decreasing the histones octamer/DNA interaction. Finally, G27A, E36G, and M63K were found in female genital tract carcinosarcomas and S37 and Y38 in follicular lymphomas.
Histone H4
Histone H4 is not as frequently mutated as other histones, nevertheless, some recurrent mutations in the globular domain and the N-Terminal region have been identified. The most frequent of these is R3C, although R3 can also be demethylated or citrullinated, resulting in deregulation that is associated with cancer. Another mutation that has been widely studied occurs at R45, which is located in the globular domain of H4. This and other single amino acid changes within H3 or H4 are referred to as Sin- (SWI/SNF-independent) mutations, where gene expression and higher-order chromatin folding become independent of the SWI/SNF remodeling complex. Finally, D68 and R92 mutations may also play a crucial role in H2B/H4 interaction as their mutation also leads to nucleosome instability.
What Are the Consequences of Histone Mutations in Cancer Development?
H3K27M in Diffuse Intrinsic Pontine Glioma
Diffuse Intrinsic Pontine Glioma (DIPG) is an aggressive form of pediatric high-grade glioma (pHGG) with a 5-year survival rate less than 1%. H3K27M is found in 30% of pHGG and 80% of DIPG, and it is the only one recognized by the WHO as a marker for tumor classification. Given the low survival and the aggressiveness of DIPG, looking for new therapy is a priority. In 2015, a collaborative study was published in Nature Medicine where several cancer drugs were tested in vitro in DIPG patient tumors and in vivo in mouse DIPG xenografts. 83 drugs were tested in culture and showed that HDAC inhibitors were the most efficient. The drug candidate Panobinostat, a multi-HDAC inhibitor, was chosen for further investigation. Panobinostat reduced DIPG cell survival by limiting proliferation and increasing apoptosis. Interestingly, drug treatment increases K27 trimethylation and acetylation, which is consistent with a previous study showing that H3K27ac can counteract PRC2 inhibition by K27M. RNA-Seq revealed “back-to-normal” transcriptome with the reversion of the K27M gene signature. The effect of Panobinostat was further confirmed in DIPG orthoptic xenograft. The treatment slows down the tumor growth and increases survival. DIPG can display resistance to Panobinostat chronic treatment, however, Panobinostat can synergize with GSKJ4 (demethylase inhibitor) in H3.3K27M by decreasing cell viability. The study opens the door to potential new treatments for this incurable cancer.
Oncohistones and Nucleosome Stability
H2B E76K is a mutation found at a high frequency in cancer. It is located in the globular region of H2B and has a dominant-negative effect on the wild-type form of the protein. Arimura Y. et al. analysed the effect of H2B E76K on the nucleosome. They first compared in vitro E76K-containing nucleosome to wild-type nucleosome. Because of the electrostatic repulsion of the H2B side chain, the H4 α3-helix displays drastic changes in the configuration leading to H2B/H4 interaction modification. The perturbation of the interaction induces the instability of the nucleosome in vitro. The group further tested the effects of E76K on cells in culture. As compared to control, the cells expressing H2B E76K formed more colonies, without impacting the cell cycle progression. E76K can induce an oncogenic-like phenotype. The H3 E97K seems to have the same effect as the H2.B E76K to a lesser extent. Mutations in the globular region of histone tend to induce nucleosome destabilization, however. the mechanism underlying the colony formation increase still needs to be explored.
Core Mutations: Functional Study
K27, G34, and K36 in histone H3.3 are the most studied histone mutations in the literature. To analyze the consequences of these mutations, Bagert JD. et al. built a DNA-barcoded mononucleosome library with canonical and variant histones, mutated or not. They used it in vitro and also in yeast. Most of the mutants tended to increase dimer exchange. The most destabilizing mutations were located at the dimer/tetramer interface between H2B/H4, including H2B, D68, F70, E71, and E76. The remodeling rate was also increased with mutations in residues in contact with DNA, such as H2AR29, K74, K75, and R77, H2BG53, and H4R45. They further analyzed the effect of these mutations on transcription in C3H10T1/2 progenitor cells. H2B mutant cell lines showed several signaling pathways and cell adhesion defects, including PI3K-Akt, Wnt, and Rap1 pathways. Interestingly, pathways involved in pluripotency and cancer-associated proteoglycans were also upregulated. Finally, when they submitted cells expressing E71K/Q, E76K/Q, and E113K/Q to adipocyte and chondrocyte differentiation, they observed that charge-swap mutation (K substitution) inhibits cell commitment, contrary to charge-neutralizing mutation (Q substitution) and wild-type histones. Altogether, the bar-coded nucleosome library allows the functional analysis of oncohistones and the mutation effect on tumor progression.
Summary: Understanding the Variety of Oncohistone Types as Tools in Drug Discovery & Personalized Medicine
Oncohistones are found in many different kinds of cancer, in children, adolescents, and adults. The mutations found in oncohistones are located in the N-Terminal tails as well as in their globular domains. The consequences of these mutations are not all known but some of them are already shown to be associated with cancer acceleration and aggravation.
Scientists are already taking advantage of this knowledge to test drugs either targeting the mutation or its consequences. The cancer-type specificity of oncohistones could help in development of targeted therapy, while limiting the side effects that occur with chemotherapy. Methods to detect these mutations could also help in diagnostic and prognostic applications and further the efforts toward personalized medicine. Lots of work still need to be done but the study of oncohistones paves the way towards potential new therapies. Stay tuned!
About the author

Anne-Sophie Ay-Berthomieu, Ph.D.
Anne-Sophie was born in the south of France and grew up between the Mediterranean Sea and the Pyrenean Mountains. She grew up as a science fiction fan, leading her to specialize in molecular biology and genetics during graduate school at the University of Lyon, France (secretly hoping her research would give her superpowers!). After living in different places for work, she is back in Lyon, France where she shares her time between her husband, her family, and her friends. During her free time, Anne-Sophie challenges herself with hiking, climbing, racing, and traveling in foreign countries – while waiting for her superpowers to grow!
Contact Anne-Sophie on LinkedIn with any questions, or to tell her about your superpowers.
Related Articles
Epigenetics of the Indian Cobra: Multi-Omics Breakthroughs for Recombinant Antivenom

May 11, 2021
A study which combines elegant, high-throughput, and cutting-edge omics helps to assemble the Indian cobra genome and may aid in the creation of an effective recombinant antivenom.
Read More
Complete Guide to Understanding Single‑Cell RNA‑Seq
March 4, 2021
Single-cell RNA-seq techniques have made it possible to study transcriptomics in heterogeneous samples; driving advances in our understanding of cancer, embryonic development and neurodegenerative disease. This article covers the history, protocols, and applications of Single-cell RNA-seq.
Read More
<< Back to MOTIFvations Blog Home Page



