<< Back to MOTIFvations Blog Home Page
低コストでハイスループットな 3’-Digital Gene Expression (3’-DGE) RNA-Seq
June 14, 2022
目次
はじめに
10年以上に渡り、RNAシーケンス (RNA-Seq) は、個々の細胞や細胞集団内の全RNA転写産物「トランスクリプトーム」において、解像度の高い情報を提供する有用な技術となっています。トランスクリプトームのデータは、転写制御、細胞分化から発がんに至るまで、多くのプロセスに関する知見を深めてくれます。
RNA-Seqの成功は、RNAからの高品質なライブラリー調製にかかっています。これまで多くのライブラリー調製法が開発されてきましたが、どの方法も基本的には、RNAの断片化、断片化RNAからの相補的DNA (cDNA) 変換、アダプター付加、そして次世代シーケンスに必要な量のライブラリー増幅といったステップで構成されています。また、ライブラリー調製にかかる時間とコストの削減にも多くの挑戦がなされましたが、必要なシーケンス深度は変わらず、2,000万~3,000万、もしくはそれ以上のペアエンドリードが必要なままでした。シーケンスコストは徐々に下がっているものの、数百サンプルとなるとやはり高額になります。
あまり知られていませんが、高品質な遺伝子発現解析データを保ちながら、シーケンス深度を大幅に浅くできるライブラリー調製法があります。それは「3’-Digital Gene Expression (3’-DGE)」という手法で、わずか300万~1,000万のシングルエンドリードしか必要としません。全てのRNA-Seq用途に適しているわけではありませんが、正しく使用すれば、発現量の違いや定量的なトランスクリプトーム解析の結果を、従来法よりもはるかに低コストで得られます。
3’-Digital Gene Expression (3’-DGE) RNA-Seqとは?
大量のRNA-Seqをハイスループットに行う目的として、トランスクリプトームアセンブリ、選択的スプライス部位や転写産物アイソフォームのキャラクタライゼーション、バリアント検出やアレル特異的発現解析などがあります。ですが、RNA-Seqの主な目的は、様々な実験条件によってアップレギュレーションまたはダウンレギュレーションする遺伝子とそれらの経路の特定です。
多くのRNA-Seq実験方法のうち、3’-Digital Gene Expression (3’-DGE) は、転写産物の3’末端をターゲットとする、遺伝子発現量の違いを解析するために最適化された新しいRNAシーケンス法です。3’-Digital Gene Expressionという単語の「Digital」は「数える」という意味です。そのためこの方法は、「transcript counting」法とも呼ばれます。
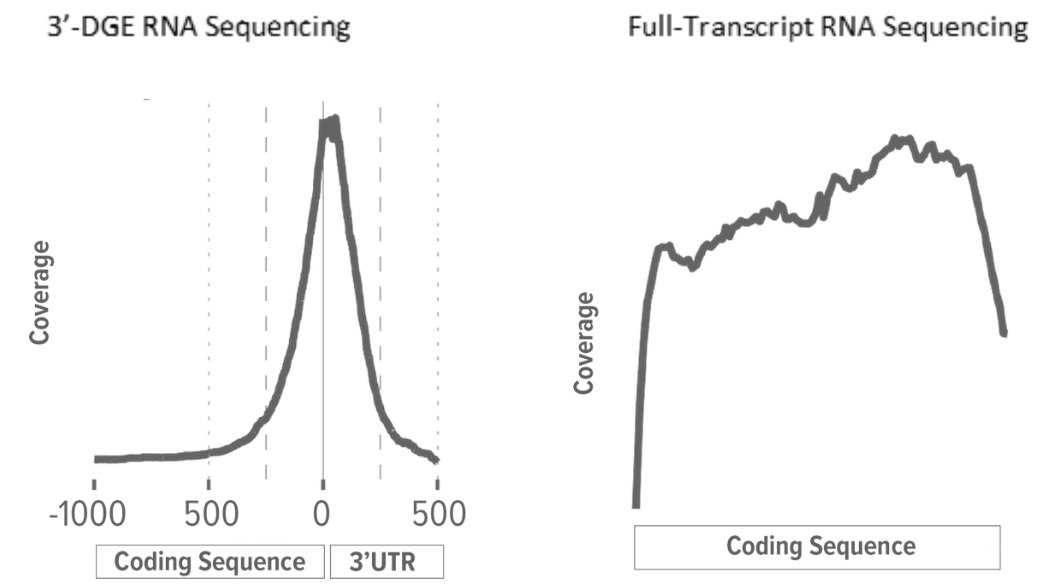
Full Transcript RNA-Seqと3'-DGE RNA-Seqの比較
従来のFull Transcript (FT) RNA-Seqは、ランダムなRNA断片化と、cDNA合成ステップでのランダムなプライミングにより、発現したトランスクリプトーム全体をマッピングするシーケンスリードを生成します。その結果、参照トランスクリプトームまたはゲノム配列にマッピングされたときに、発現する全転写産物のエクソン全域に対応するシーケンスリードが得られます。従来のRNA-Seqにおいて一つの遺伝子にマッピングされるリード数は、転写産物の量と長さの関数となります。つまり、長い遺伝子は、転写産物量が同程度の短い遺伝子よりも、マッピングされるリード数が多くなります。
3‘-DGE RNA-SeqライブラリーのcDNA合成ステップでは、ランダムプライマーを使用せず、オリゴ (dT) 付きアダプターを使用して、転写産物 (mRNA) のポリAテールをプライミングします。そのため、3‘-DGEのリードは、転写産物の3’末端にマッピングされる傾向があります。転写産物とcDNA分子の間には理論上の1対1の相関関係があるため、転写産物の長さに関係なく、転写産物の存在量に比例します。つまり、短い遺伝子と長い遺伝子で発現量が同程度であれば、マッピングされるリード数も同程度になります。

3'-DGE RNA-Seqの利点と弱点
産物のポリAテールへのプライミングに依存しているため、真核生物にしか使えません。次に、 3‘-DGEリードの大部分は遺伝子の3’-UTRにマッピングされるため、トランスクリプトームのアノテーションが充実していることが望ましいと言えます。ただ、これには回避策があります。翻訳停止部位が明らかにもかかわらず、転写停止部位がアノテーションされていない場合、バイオインフォマティクス的にリファレンスシーケンスを作成、もしくは対象の領域を、翻訳停止部位から上流や下流の距離に基づいてアノテーションする、といった方法です。ストランド特異的RNA-Seqであれば、逆ストランド上の隣接遺伝子の3’-UTR付近にマッピングされたリードを区別できるため、3‘-UTR長の過剰な予測による問題を回避できます。3つ目は、解析の柔軟性の問題です。転写産物全体からのデータを必要とする解析 (アセンブリ、スプライシングなど) の場合には、従来法のRNA-Seqを使用するか、 2 つのアプローチを戦略的に組み合わせて使用することをお勧めします。これらの弱点の他、3’-DGEがRNA-Seqに利用されない主な理由には、単純に手法そのものと、利点がよく知られていないことがあるでしょう。
3’-DGEでの転写産物とcDNA分子の1対1の相関関係は、transcript countingを効率化し、データ解析を簡単にします。例えば、遺伝子Aにマッピングされる100リードのRNA-Seqサンプルを考えてみましょう。従来のRNA-Seqでは、これら100リードの基になった転写産物がいくつあるのか、はっきりとしませんでした。この問題は転写産物の長さを正規化しても変わりませんが、3‘-DGEでは原則として100リードが100分子の転写産物に相当します。
解析が簡単な点は、3‘-DGE法の最も大きなメリットの一つです。Transcript countingは、従来のRNA-Seqよりも浅いシーケンス深度で、確実な解析ができることを意味しています。また3’-DGEには、ペアエンドシーケンスが不要です。従来のRNA-Seqライブラリーを1サンプルあたり2,000万~3,000万のペアエンドリードでシーケンスしていた場合、同じサンプルを3’-DGEライブラリーにすることで、1サンプルあたり300万~1,000万シングルエンドリードでシーケンスできるため、大幅にコストを削減することができます。予算の関係でRNA-Seq実験のレプリケーションを諦めていたなら、安価で確実な3’-DGEを利用することでレプリケーションを増やすことができるため、統計的でより正確な遺伝子発現解析が可能になります。

3'-DGEの利用で得られる発見
RNAの扱いには注意が必要です。目に見えないRNaseの存在は、RNA抽出を初めて (たとえ100回目でも!) 行う研究者を悩ませ、夜も眠れなくします。抽出したRNAが高いRIN (RNA Integrity Number) 値を示せば安心できますが、サンプルサイズが大きくなるほど、RNAの品質にばらつきが生じるのは珍しくありません。全転写産物の状態に影響を受ける従来のRNA-Seqライブラリーでは、RNA品質のばらつきは解析上の大きな問題となります。サンプルの種類によっては、どのようなRNA抽出法を使っても高品質なRNAの抽出ができず、従来のRNA-Seqでは解析できないことがあります。しかし、RNA品質のばらつきによる影響が小さい3‘-DGEライブラリーを用いることによって、このようなサンプルについても遺伝子発現解析が可能になるという利点があります。
バリアント検出は、個体群をシーケンスする場合は特に、包括的RNA-Seq解析に新しい一面をもたらす可能性があります。タンパク質をコードするエクソンは、遺伝子の非翻訳領域よりも大きな選択圧を受けていることを考えると、3‘-DGEでは従来のRNA-Seqよりもバリアントに関するリードが多く得られるはずです。このバリアント情報は、たとえば戻し交雑自殖系統/準同質遺伝子系統: BILs/NILs (backcross inbred lines/near isogenic lines) やゲノムワイド関連解析: GWAS (genome-wide association study)、量的形質遺伝子座: QTL (Quantitative trait locus) のマッピングなど、遺伝子発現研究以外の解析にも有用です。この低選択圧と3’-UTRを利用して、鳥類の系統樹を解明したという論文も出ています (論文はこちら)。
アノテーションが充実している真核生物のゲノムや、転写産物のスプライシング情報が不要なRNA-Seqの実験には、3’-DGEの利用をお勧めします。この方法はシーケンスコストの半減、簡単なデータ解析、そしてRNA品質の影響を受けにくいなど、多くの利点があります。3’-DGEシーケンスを始めるなら、アクティブ・モティフのYourSeq (FT & 3’DGE) Strand-Specific mRNA Library Prep Kitをお試しください。
About the author

Mike Covington
Mike was co-founder and CTO of Amaryllis Nucleics, an RNA-Seq company that recently joined Active Motif. Before transitioning to bioinformatics and next-gen sequencing, Mike started his career at the bench as a molecular biologist studying the physiological relevance of the plant circadian clock. As an open-source science and software enthusiast, he has spent the last decade focused on developing bioinformatic tools, science-related web apps, and new molecular biology tools. A native of the Pacific Northwest, Mike has spent most of his life up and down the West Coast where he enjoys adventures in/on the water, experimenting with music and languages, reading with a cat on his lap, and creating vegan junk foods.
Related Articles
Library QC for ATAC-Seq and CUT&Tag AKA “Does My Library Look Okay?”
December 8, 2021
“Does my library look okay?” is the most common question posed to Active Motif technical support. Get the practical scoop on quality control for ATAC-seq and CUT&Tag libraries. Find out how much library to expect from different libraries, what they should look like, and what to do if it’s not as expected.
Read More
PROTAC-mediated Targeted Protein Degradation in Cancer: PARP, EGFR, and SMARCAs in Focus
April 13, 2022
Proteolysis-Targeting Chimera (PROTAC) protein degraders are the emerging alternative to small molecule-based targeting. Here we look at key protein targets and discuss how PROTAC-mediated targeted protein degradation represents a promising new approach to cancer treatment.
Read More
<< Back to MOTIFvations Blog Home Page



