<< Back to MOTIFvations Blog Home Page
完全ガイド:ChIPアッセイにおけるクロマチンのソニケーション

By Anne-Sophie Ay-Berthomieu, Ph.D.
January 31, 2020
クロマチン免疫沈降法(ChIP)は、DNA結合タンパク質およびそれに関連するDNA配列を解析するための最も標準的な手法です。この手法は、転写因子やコファクター、さらにはヒストンやヒストンの翻訳後修飾を研究するために広く用いられています。
しかし、ChIPは多くの工程を要する実験であり、良好な結果を得るためには各ステップを慎重に行う必要があります。ほとんどのプロトコルでは、まずサンプルを架橋・断片化し、次に目的のタンパク質またはヒストン修飾に特異的な抗体を用いてクロマチンを免疫沈降し、その後、免疫沈降されたDNAを次世代シーケンシングやリアルタイムqPCRアッセイを用いて解析します。
クロマチンの断片化はChIPワークフローにおいて最も重要なステップの一つであり、一般的に最もばらつきが生じやすい工程でもあります。断片化(シアリング)には酵素的方法と機械的方法(ソニケーション)があります。ソニケーション(超音波処理)はプローブ型ソニケーター、カップホーン型ソニケーター、ウォーターバス型ソニケーター、または焦点型ソニケーターが用いられます。
それぞれのシアリング手法には、その利点と限界があります。本記事では、各ソニケーション法の違いについて解説し、それぞれの長所と短所を明らかにします。
全てはクロマチン固定から始まる
タイトルの通り、ほとんどの場合、固定化が最初のステップです。固定化は、対象となるタンパク質をクロマチンに架橋させ、クロマチンと動的に、あるいは低親和性で結合している可能性のある因子を解析できるようにするために行われます。
固定条件を最適化する際に考慮すべきおもな基準は、使用する固定剤(一般的にはホルムアルデヒドを使用します)、固定剤の濃度、固定時間、固定温度、および固定の停止方法です。
使用する細胞や組織の種類、対象とするエピトープに応じて固定条件が異なる場合があるため、最良の結果を得るには、サンプル種やChIPのターゲットごとに固定条件を最適化することをお勧めします。
固定プロトコルの最適化法
ホルムアルデヒドは、粉末(パラホルムアルデヒド)または溶液(ホルムアルデヒドまたはホルマリン)の形で入手できます。パラホルムアルデヒドはホルムアルデヒドが重合した粉末であり、それ自体には固定作用はありません。固定剤として使用する場合、水に溶解してホルムアルデヒド分子を遊離させる必要があります。
ホルムアルデヒドは溶液中では不安定であり、その固定能力は時間経過とともに低下します。このため、一部のホルムアルデヒド溶液には安定化のためにメタノールが含まれています。しかし、メタノール自体にも固定作用があるため、固定プロトコルの最適化においてはこれを考慮する必要があります。再現性のある結果を得るための最良の方法は、ホルムアルデヒド溶液(ホルムアルデヒド濃度は1%以下)を用時調製すること、またはメタノールを含まない使い捨てアンプルを使用することです(訳注:歴史的には、ホルムアルデヒド溶液に安定剤として含まれるメタノールが固定条件に影響を与えると考えられていましたが、ChIPアッセイで用いるホルムアルデヒド濃度は1%程度であり、ここまで希釈するとメタノール濃度は0.5%未満となり、架橋にあたえる影響は非常に少ないと考えられます)。
最適な固定時間および温度は、使用するソニケーターの種類、細胞・組織のタイプ、そして標的タンパク質によって異なります。プローブ型およびウォーターバス型のソニケーターは、高エネルギーの超音波を照射する一方で、処理中のサンプル温度を制御できません。これらのソニケーターを用いる場合にも、ソニケーション中に標的エピトープが維持され、免疫沈降に用いる抗体が認識可能な状態を保つ必要があります。そのため、十分な固定時間を確保し、氷上ではなく室温で固定を行います。
固定時間は2分~30分の範囲ですが(ほとんどのプロトコルでは10~15分を推奨しています)、これはサンプルタイプや標的タンパク質ごとに検証する必要があります。また、固定時間を正確に一定とするため、固定終了のタイミングで冷やしたグリシン溶液を添加します。これによりホルムアルデヒドによる架橋反応が停止します。
ホルムアルデヒドは最も一般的な固定剤ですが、これに併用して、あるいは代替としてより長いスペーサーアームをもつ他の固定剤を使用することもできます。特に、DNAに直接結合しない因子の解析を行う場合、ジメチルアジピミデート(DMA: dimethyl adipimidate)や、ジメチル3,3‘-ジチオビスプロピオンイミデート(DTBP: dimethyl 3,3’-dithiobispropionimidate)、グルタル酸ジスクシンイミジル(DSG: disuccinimidyl glutarate)などの他の固定剤を用いるプロトコルがいくつか報告されています。

固定不足や過固定がもたらす影響とは?
サンプルの固定が不十分な場合、単純にタンパク質とDNAの相互作用が失われることがあります。これは特に、DNAと直接結合しないコファクターや、DNAと一過性に結合する因子や、DNAとの親和性が低い転写因子を調べる実験において問題となります。
一方で、過度の固定化は、さまざまな問題を引き起こすことがあります。例えば、対象となるエピトープが損なわれ、ChIPで検証済みの抗体でさえ、標的タンパク質を認識できなくなります。また、過固定はクロマチンの断片化を困難にし、再現性のある断片長を得られなくする恐れがあります。つまり、これらの問題を回避するためには、適切な固定条件の最適化が不可欠です。
Native ChIPの仕組みと特徴
多くのChIPプロトコルでは、タンパク質とクロマチンの相互作用を調べるために、最初のステップとしてサンプルを固定します。しかし、状況によっては固定を行わないNative ChIP (N-ChIP)と呼ばれるアプローチが適している場合があります。
N-ChIPアッセイの手順は、固定の工程を除けば、従来のプロトコルとほぼ共通しています。DNAがヒストンに巻き付いた構造(ヌクレオソーム)は安定した複合体であるため、N-ChIPはヒストンの翻訳後修飾の解析に適しています。また、結合が非常に強固な、あるいは高密度に存在する一部の転写因子にも有効な場合があります。
多くの標準的なN-ChIPプロトコルでは、細胞や組織の回収、あるいは核の単離直後に、ミクロコッカスヌクレアーゼ(MNase: Micrococcal Nuclease)処理によりクロマチンを断片化します。この手法は、標準的なプロトコルよりも簡便かつ短時間で済むだけでなく、ほとんどの抗体は未固定(未変性)の抗原に対して作製されているため、標的に対する親和性を高められる可能性もあります。また、固定サンプルを用いた手法に比べてクロマチンの回収率やモノヌクレオソームの収量が増えるため、少ないサンプル量の場合でもN-ChIPの方が効率的に機能することがあります(SA David et al., 2017, J. Brind’amour et al., 2015)。
加えて、固定しないことにより細胞は自然な状態に保たれ、架橋反応の副産物として生じるアーティファクトを回避できるため、より生物学的に妥当な結果が得られると期待されます。固定作業が不要な分、ホルムアルデヒド固定を用いるプロトコルよりも迅速で、ChIPを初めて扱う研究者にとって魅力的な選択肢になります(※訳注:後述のCUT&RUNやCUT&Tag法では固定を行わないだけでなく、N-ChIPよりもさらに簡便化された方法となっています)。
以上がN-ChIPの大きな利点ですが、現実にはこの手法がうまく機能するのはごく一部の実験条件に限られます。解析対象のタンパク質が動的あるいは一過的にクロマチンと相互作用している場合、または結合の親和性が低い場合では、一般的にシグナルを得るために架橋が必要です。また、未固定条件では酵素処理中にヌクレオソームが別のDNA領域に再配置してしまい擬陽性となることもあるため、ヒストンやヒストン修飾のゲノム上の正確な位置を調べる実験にN-ChIP法を用いる場合にはリスクが伴います。
最良の結果を得るために:クロマチン調製プロトコルへの核単離の導入
ほとんどのChIP実験の目的は、核内タンパク質とゲノムDNAの結合を解析することにあります。そのため、ChIPアッセイにおいて最も感度が高く特異的な結果を得るには、実験プロトコルに核単離の工程を取り入れるべきです。
ChIP前に行う核単離の手法
固定、反応停止、洗浄を終えた細胞ペレットから、ペッスルとガラス管で構成されるダウンス型ホモジナイザー(Dounce homogenizer or Douncer)を用いて核を単離することができます。精密に設計されたペッスルを上下させることにより細胞に剪断力を与え、細胞膜を破壊しつつ、核やオルガネラを完全な状態で取り出すことができます。この器具を用いた手法は、効率的かつ低コストで核を単離可能な方法であり、後続のChIP反応を阻害するような特殊なバッファーも必要としません。しかし、時間がかかる上、サンプル間や実験間の再現性を確保するのが難しく、大規模なサンプル処理に不向きという欠点もあります。
また、ショ糖密度勾配を利用して細胞質と核を分画し、クロマチンを高純度で単離することも可能です。このアプローチでは、低速遠心により核をペレット化させる一方、細胞質画分を上清として取り除きます。さらに、より迅速かつ簡便な方法として、1% Triton X-100を含むバッファーで細胞を溶解して核を回収する手法もあります。この条件下ではクロマチンが不溶性となるため、遠心により回収できます。
最後に、2016年に発表されたNEXSON (Nuclei EXtraction by SONication)という手法を紹介します。これは超音波処理を用いて効率的に核を単離する方法です。NEXSONはおもに2つのステップで構成されており、まず固定した細胞ペレットを核抽出用のバッファーに再懸濁し、次いで適度なソニケーションを行うことにより細胞膜を破壊し、核を放出させます。この手法を成功させるには、超音波の処理時間や出力の最適化が必要ですが、開発者らによると、他の核単離法よりも高品質で再現性の高いChIP-Seqデータが得られると報告されています。
ChIPアッセイに核の単離は必須か?
前述の通り、ChIP-SeqやChIP-qPCRのおもな目的は、核内におけるタンパク質とゲノムDNAの相互作用を解析することです。真核生物では、タンパク質は細胞質で合成された後、その一部が核内へと輸送されます。核を単離しない場合、細胞質画分と核画分に局在する標的タンパク質が混在した状態となります。この状態で免疫沈降を行うと、細胞質に局在する標的タンパク質にも抗体が結合してしまい、核内の標的タンパク質に結合すべき抗体が奪われてしまう可能性があります。そのため、最終的なシーケンシングやqPCR解析において検出シグナルを低下させるリスクを上昇させます。
一方で、核の単離は手間がかかり、初心者にとっては再現性が得られにくいこともあります。また、工程が増えることによるサンプル損失を招く可能性もあるため、サンプル量が非常に少ない場合では、ChIP反応に十分な量のクロマチンが残らないというリスクも生じます。以上のように、核の単離はChIPアッセイの質を向上させますが、サンプル量が限られているケースなどではむしろデータ品質を低下させる要因にもなり得るため、必ずしも必須とまでは言えません。
なお、PIXUL® Multi-Sample Sonicatorのような最新の装置では、細胞や組織サンプルを単一のバッファーで処理することが可能です。PIXULを用いて断片化したクロマチンによるデータでは、核を単離せずとも高品質な結果が得られることが実証されています。
PIXUL Multi-Sample Sonicatorについての詳細は、弊社のPIXUL-ChIPに関する記事をご覧いただくか、以下のポッドキャスでPIXUL発明者のKarol Bomsztyk医師およびTom Matula博士へのインタビューをお聴きください。
最良のクロマチン断片化法とは?
クロマチンの断片化はクロマチン免疫沈降法において最も重要な工程の一つであり、細胞や組織の種類、さらに標的タンパク質に応じて慎重に最適化する必要がります。また、クロマチンを断片化する方法はいくつかありますが、それぞれに長所と短所があります。
ソニケーション法を用いた機械的なシアリング
ソニケーションの仕組みと特徴
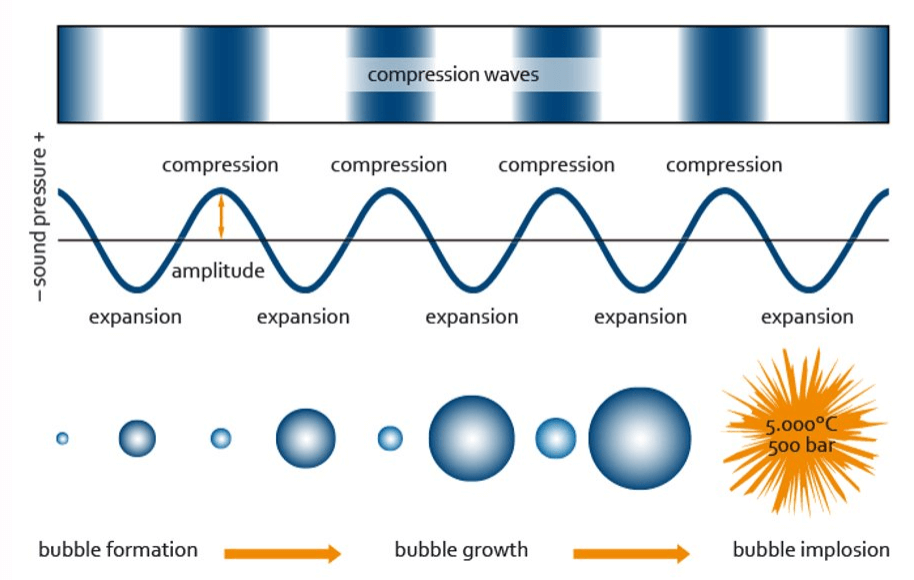
ソニケーターは電気エネルギーを超音波エネルギーに変換し、それを処理対象のサンプルに伝達します。ソニケーターにより生成される音波は、音の周波数に応じて、圧縮と膨張のサイクルを交互に繰り返します。膨張サイクル(低圧期)において、高強度の超音波は微小な空洞(キャビティ) を発生させます。これらの泡状の空洞がそれ以上エネルギーを吸収できなくなると、次の圧縮サイクル(高圧期)で激しく崩壊します。この現象はキャビテーション(空洞現象)と呼ばれ、この崩壊時に生じる強力な剪断力がソニケーション中にDNAやクロマチンを断片化させます。
1サンプルずつ処理するプローブ型ソニケーター
プローブ型ソニケーターは、一度に1つのサンプルを処理するシンプルな装置です。比較的安価であるため、ほとんどの研究室で導入しやすいという利点があります。しかし、プローブ型を使いこなすには熟練を要し、科学というよりはむしろ「職人技」に近い側面があります。
プローブ型を使用する際のもう一つの留意点は、冷却システムが統合されていないことです。そのため、通常はアイスバケットなどを使ってサンプルを冷却しながら処理を行います。しかし、アイスバケット内の氷の状態は一定ではないため、試行ごとにサンプルの温度変化のパターンに差が生じ、それが不安定な結果の原因となることがあります。サンプル間の再現性を高めるため、低温を維持してプローブを常に同じ深さに保持できる冷却システムも存在します。
また、プローブ型にはサンプル間のコンタミネーションのリスクも伴います。サンプルを替える際にプローブを軽く洗浄するだけなため、前のサンプルに含まれる物質が次のサンプルに持ち込まれる可能性があるためです。さらに、処理中のチューブはプローブを挿入するために開放されていることが多く、チューブ内の有害物質がエアロゾル化する危険性もあります。
そして、おそらく最も重要な点として、プローブ型を用いて多検体の実験を行うには非常に時間がかかり、シアリングの再現性を確保することがしばしば困難になります。
ハイスループットなクロマチン断片化を実現するマルチサンプルソニケーター
マルチサンプルソニケーターは、複数のサンプルを同時に処理することができます。これらの装置はウォーターバス型や焦点照射型の方式を採用しており、冷却システムも統合されています。このタイプの装置を用いることにより、再現性と効率性、そして正確な温度管理が保証されます。ただし、マルチサンプルソニケーターの中には非常に高価なものもあり、特に市販されている一部のハイスループットソニケーター専用のプレートは、1枚で15万円以上する場合もあります。
各種マルチサンプルソニケーターの具体的な詳細については、後述します。
酵素を用いたクロマチンの断片化
酵素によるクロマチンの断片化では、通常、ミクロコッカスヌクレアーゼ(Micrococcal Nuclease, MNase)を用いてクロマチンを小さな断片へと消化します。MNaseはエキソヌクレアーゼ活性とエンドヌクレアーゼ活性の両方を有しており、DNAを最小でヌクレオソーム1個分(約147bp)のサイズまで消化することが可能です。特別な装置を必要とせず、安価かつ迅速に処理できるという点から、酵素による断片化を好む研究者もいます。また、酵素消化はソニケーションほど物理的刺激が強くないため、解析対象のエピトープが保護され、抗体による認識が維持されやすいというメリットがあります。
しかし、MNaseによる消化には特定のバイアスが存在します。MNaseは、GC含量の多い配列よりもAT含量の多い配列を切断しやすいことが示されています。また、ヘテロクロマチン領域(高度に凝集した領域)は酵素がアクセスしにくいため、消化パターンにさらなる偏りが生じます。このように、酵素を用いたクロマチン断片化では非ランダムな切断となるため、実験目的に応じてその特性に留意して使用する必要があります。
プローブ型ソニケーターを使用する際の重要な留意点

プローブ型ソニケーターを使用する場合、プローブをサンプルにじかに挿入して「直接ソニケーション」を行います。この方式はエネルギー伝達効率が非常に高く、クロマチンをシアリングするのに十分な強度を得ることができます。プローブ型でクロマチンを処理する際は、通常、0.1%~10%のSDS (Sodium Dodecyl Sulfate:ドデシル硫酸ナトリウム)や、プロテアーゼおよびフォスファターゼ阻害剤を含むバッファーでサンプルを希釈します。SDSはソニケーションの効率やクロマチンの収量を高めるだけでなく、エピトープを露出させて抗体が認識しやすくする効果があります。しかし、SDS濃度が高すぎると、DNAと直接結合していないタンパク質との相互作用を阻害したり、結合を失わせたりする可能性があるため注意が必要です。
プローブ型ソニケーターは非常に高いエネルギーを供給するため、サンプルの過熱を防ぐために低温に保たなくてはなりません。そのためにはアイスバケットや、より効果的な冷却ラックなどを使用します。ただし、氷は常に一定の低温を維持できるわけではなく、複数のサンプルを処理している間に溶けてしまうこともあります。最良の方法は、ソニケーションの全工程、およびサンプル間において一定の温度を保つような冷却システムを使用することです。
さらに、プローブを用いてソニケーションを行う際は、プローブがチューブの側面に触れないようにすること、そしてサンプル間のシアリング状態に一貫性を持たせるために、異なるサンプル間でもプローブを同じ深さに挿入することが重要です。複数のサンプルを連続して処理する場合は、クロスコンタミネーションを避けるために、サンプルごとにプローブを注意深く洗浄することも重要です。
最良の結果を得るためには、パルス数、パルス持続時間、およびパルス強度をすべてサンプルの種類やChIPのターゲットごとに最適化しなければなりません。最適化の基準としては、例えば25%の強度設定で5回、10回、20回のパルスを試してみるのが良いでしょう。この時、20秒間のソニケーションと、サンプルを低温に保つために30秒間の休止で構成します。
核の単離が行われていない場合や、クロマチンが非常に高度に凝集している場合は、効率的なソニケーションを行うために、おそらくソニケーション時間や強度の設定値を大きくする必要があるでしょう。逆に、解析対象のタンパク質が分解されるリスクがある場合は、ソニケーション時間や振幅を抑える必要があります。
多検体のソニケーションを行う際の重要な留意点

ほとんどの多検体用ソニケーターは、ウォーターバス型(例:Bioruptor® PICO)または焦点照射型(例:Covaris®)のいずれかを採用しています。プローブ型ソニケーターとの比較において、多検体用のおもな利点は、シアリングの再現性とクロスコンタミネーションの排除、そして手作業の時間を減らした迅速な処理です。プローブ型とは異なり、多検体型は通常、冷却システムが装置に組み込まれているため、サンプル間における処理の均一性が向上します。
ウォーターバス型のBioruptor Picoは1回のランで最大16サンプル、焦点照射型のCovarisは最大96サンプルの同時処理が可能です。しかし、これら2つの装置はソニケーション方法が異なります。Covarisで採用されている焦点型超音波では、各サンプルの下に配置されたトランスデューサーが超音波を発生し、生成されたエネルギーが照射対象のウェル(またはチューブ)に確実に集中するように設計されています。対照的に、ウォーターバス方式を用いるソニケーターでは、超音波が液体中をランダムに伝播するため、エネルギーが分散してしまいサンプル間に不均一性が生じやすくなります。ウォーターバス型ソニケーターには通常、超音波強度の強い『ホットスポット』が存在します。そのため、これらの装置では、すべてのサンプルが少なくとも一定時間はホットスポットに収まるようにサンプルを回転させる機構が用いられます。
一般に、ウォーターバス型ソニケーターでは比較的低い周波数の超音波を用いるため、細胞をやクロマチンを効率的にシアリングするにはより多くのエネルギーが必要となります。その結果、サンプルに熱がたまりやすくなります。ソニケーションにおいて過度な温度上昇は常に注意すべきであり、DNAや目的のタンパク質にダメージを与えるおそれがあります。焦点照射型ではより高エネルギーの高い周波数の音波を用いるため、より効率的にエネルギーを伝達可能となり、処理時間の短縮と発熱の抑制が可能です。
ほとんどの多検体用ソニケーターでは、使用前に冷却水の脱気を行うことが極めて重要です。水中の気泡や不純物は音波を散乱し、ソニケーション効率を低下させる原因となるためです。脱気に要する時間は装置によって異なり、数時間を要することもあります。
多検体用ソニケーターを使用する際には、消耗品のコストも重要です。汎用のチューブを使用できる装置もあれば、専用かつ高価なプレートやチューブにしか対応しない装置もあります。

既存の多検体用ソニケーション技術にはそれぞれ大きな制約があるため、Active MotifはMatchstick Technologiesと共同で、PIXUL Multi-Sample Sonicatorを開発しました。
PIXULは複数のトランスデューサーとレンズのアレイを用いて超音波エネルギーを集束し、比較的安価な汎用の96ウェル培養プレートを用いて、最大96サンプルを同時に処理できます。細胞は同じプレート上で培養から固定、ソニケーションまで行えるため、プレート間で内容物の移し替えが不要となり、サンプルが目減りするリスクもありません。そのため、PIXULはハイスループットなクロマチン断片化に適した装置といえます。
PIXULの冷却システムは装置に完全に内蔵されており、各ランの開始前に専用冷却液を10〜15分冷却するだけで使用でき、脱気は不要です。
PIXUL Multi-Sample Sonicatorは、 1回のランで12種類のソニケーションプログラムを適用できるため、1回のランで複数種類のサンプルを同時に処理する用途や、難しいサンプルに対する処理条件の最適化にも適しています。
Nucleic Acids Research誌に掲載された論文において、 PIXULの技術はCovaris、およびBioruptorと比較されています。この報告では、PIXULで処理したサンプルは、CovarisまたはBioruptorで処理した同一サンプルよりも、よりばらつきの少ない断片化効率と再現性を得られることが示されました。さらに、PIXULではウェル間のクロスコンタミネーションは認められず、PIXULで断片化したクロマチンは、ChIP-qPCRおよびChIP-Seqにおいて、他の装置で断片化したクロマチンと同等、またはそれ以上の高品質な結果を示しました。

酵素的断片化の使いどころ
酵素的断片化の留意点
機械的シアリングでは核単離を省略できるのに対し、酵素的断片化ではこの工程が必須です。前述の通り、酵素的断片化はクロマチン断片化時にシーケンスバイアスを生じやすく、ヘテロクロマチンの一部領域にMNaseがアクセスしにくいため、ソニケーションに比べて効率が劣ることがあります。例えば、積極的に転写されている開放クロマチン領域にはMNaseが容易にアクセスしますが、H3K27me3のような抑制性のヒストン修飾を解析する場合は、機械的シアリングが推奨されます。ただし、ソニケーターがない場合は、ChIPアッセイに酵素的断片化を用いる例もあります。
酵素的断片化のおもな利点は、インキュベーション時間を調整することにより消化を完全に制御でき、1〜4ヌクレオソームサイズに対応した特異的なDNA断片パターンを得られる点です。ある細胞種に対してプロトコルが確立されれば、結果の再現性は非常に高くなります。
酵素的断片化は過去のものではない:CUT&RUN!
MNaseによるクロマチンの酵素的断片化にはいくつかの欠点がありますが、この酵素はChIPに類似したプロトコルであるCUT&RUN (Cleavage Under Targets and Release Using Nuclease)において改良的に活用されています。CUT&RUNでは、抗体に結合するprotein Aと融合したMNaseを用いることで、標的タンパク質に結合した抗体の近傍へ酵素を選択的に導きます。その結果、標的タンパク質が結合しているクロマチン領域のみが選択的に切断されます。CUT&RUNは通常のChIPよりもバックグラウンドが少なく、少ない細胞数でも使用可能と報告されています。ただし、ネイティブ条件(非架橋)で行うため、存在量の少ない転写因子やクロマチンと親和性の低い標的を調べる実験では検出が困難になる場合があります。
従来のChIP-Seqに代わるもう一つの方法として、CUT&Tag(Cleavage Under Targets and Tagmentation)があります。CUT&Tagは、Active Motifの特許技術であるTAM-ChIP™を基盤とした手法で、protein Aと融合したTn5トランポゼースを利用します。抗体に結合したprotein AがTn5を標的部位へ導き、標的タンパク質の近傍のクロマチンを切断すると同時に、Illumina互換のNGSライブラリアダプターを挿入します。そのため、クロマチンDNAの精製を行わずに、反応の過程で直接ライブラリを作製できます。
さまざまなサンプルタイプのソニケーションのコツ
前述のとおり、ソニケーションを含むChIPのワークフローは、最適な結果を得るために、細胞種ごと、標的ごとに各工程を検証する必要があります。
標的分子の発現量やクロマチンとの結合様式によって異なりますが、標準的な培養細胞サンプルを用いるChIPでは、一般に1,000個から500万個程度の細胞が必要です。基本的には、細胞数が多いほどChIPの成功率は高いと考えてよいでしょう。細胞によっては溶解しにくく、クロマチン収量が非常に少ないこともありますし、同じ標的タンパク質でも細胞種によって発現量が異なることもあるため、ChIPを始める際にはそれらの点も重要な考慮事項になります。
少量サンプルにおけるChIP
患者由来のサンプルや、組織生検、ソーティングした細胞集団を用いる場合、ChIPに必要なぎりぎりの細胞数しか確保できないことがあります。このような場合は、標準的なプロトコルを少し調整することで、結果を改善し、サンプルのロスを抑え、さらに全体の工程数を減らすことができます。
固定の段階でシリコンコートされたチューブを使うと、サンプルがチューブ壁に付着しにくくなるため、サンプルのロスを抑えられます。さらに、サンプル量が極めて限られている場合には、核単離の工程を省略できることがあります。免疫沈降にはChIP-Seqで十分に検証された、高感度かつ特異的な抗体を用いる必要があります。
最後に、少ないサンプル量でChIP-Seqを行う場合には、ライブラリ調製法が成功の鍵を握ります。クロマチン量が少ないChIPでは、ライブラリ調製の過程でPCR増幅を行うと重複リードが生じやすくなります。しかも、ChIPでは本来同じ座標に由来する断片が多く集まるため、配列情報だけでPCR由来の重複と、実際に濃縮されたクロマチン由来のリードを区別しにくいという問題があります。この問題に対して、Active Motifでは、アダプターにユニーク分子識別子(UMI: Unique Molecular Index)を含むNGSライブラリ調製キットを提供しており、バイオインフォマティクス解析の段階でPCR重複を除去できるようにしています。
PBMCに対するChIPアッセイ
末梢血単核球(PBMC: Peripheral Blood Mononuclear Cells)は、一般に溶解させるのが難しいサンプルです。そのため、通常の方補ではクロマチン収量が低下したり、場合によってはクロマチン品質が悪化したりします。細胞溶解を改善するには、膨潤バッファー(swelling buffer)に加えて界面活性剤を含むソニケーションバッファーを用い、上皮系の細胞株などよりもやや強めのソニケーション条件で処理することが推奨されます。
サンプルのロスを避けるため、ダウンス型ホモジナイザーを用いた核単離は推奨されません。PBMCの溶解とクロマチン断片化を十分に進めるには、やや長めにソニケーションを行う方法が適しています。PBMCに特化したChIPキットとして、アクティブ・モティフではChIP-IT® PBMCキットを提供しています。 このキットには、PBMCサンプルを効率よく溶解し、良好なクロマチン収量を得るための各種バッファーを含まれています。
FFPE組織に対するChIPアッセイ
臨床研究では、患者の臨床情報と紐づいたFFPE組織サンプルが多数蓄積されていることがあります。こうした保存検体は、過去に収集された試料を活用する研究において非常に有用ですが、パラフィン包埋ブロックから十分なクロマチン収量と良好な品質を得るのは容易ではありません。量が少ないうえに、固定・包埋条件が不明なこともあり、サンプルごとのばらつきも大きいためです。また、FFPE試料の作製に用いられる強い固定条件は、場合によっては標的タンパク質のエピトープを損なうこともあります。
FFPE組織を用いてChIPを行う際は、まずパラフィンを丁寧に除去し、組織切片を再水和する必要があります。クロマチンを抽出してシアリングするには、溶解バッファーで組織を処理し、場合によってはソニケーションに加えて酵素的断片化も併用しなければなりません。FFPE試料では過固定がしばしば起こり、クロマチンが溶解・断片化しにくくなるためです。
免疫沈降とその後のシーケンシングを開始する前に、クロマチンの品質と断片化効率を慎重に確認する必要があります。これは多くの研究者にとって大きな課題であるため、Active Motifは、FFPEサンプルから可能な限り高品質なクロマチンを抽出しやすくするために、ChIP-IT® FFPE IIキットを開発しました(販売終了しています)。
ソニケーション後のクロマチンの断片化効率と収量の評価方法
前述のとおり、ソニケーション工程は、すべてのChIP-Seqアッセイの成否を左右するきわめて重要なステップです。そのため、免疫沈降を始める前に、クロマチン収量と断片化効率を確認しておく必要があります。
脱架橋をしたら、続いてRNase AとProteinase K処理を行い、RNAとタンパク質を除去します。さらに、古典的なフェノール/クロロホルム法、あるいはより簡便な精製カラムやSPRIビーズなどを用いてDNAを抽出・精製します。精製したDNAは、最後にNanoDropのような分光光度計、またはQubit)のような蛍光光度計で定量します。
ソニケーション効率の評価には、精製したクロマチンを用いてアガロースゲル電気泳動を行う方法があります。より定量的に評価したい場合は、Agilent社のBioanalyzer、TapeStation、またはFragment Analyzerを用いてクロマチンのシアリング状態を解析することを推奨します。適切にソニケーションされた場合、DNA断片の長さはおおむね200〜600 bpになります。
ソニケーション条件を最適化する際には、標的タンパク質上の関連エピトープが、固定化やソニケーションによって失われていないかどうかを確認する目的で、免疫沈降後にウェスタンブロットを行うのも有効です。
結論:良好なソニケーションがすべてを左右する
ソニケーションは、ほとんどのChIPアッセイの成否に直結する最初の重要な工程です。もしChIPアッセイでのソニケーションが不十分であったり十分なクロマチンが得られなかったりすれば、抗体がどれほど優れていても、またその後の工程がどれほど安定していても、実験を成功に導くことはできません。良好にソニケーションがなされなければ、成功は望めません。
クロマチンのソニケーションがきわめて重要な理由は複数あります。まず、ソニケーションによりクロマチンが可溶化・遊離されるため、良好なクロマチン収量を得るにはこの工程が十分に効率的でなければなりません。また、効率的な免疫沈降と良好なピーク分解能を得るため、ChIP-SeqではDNAを適切な大きさ、すなわち200〜600 bp程度に断片化する必要があります。
ソニケーションの品質が、免疫沈降からシーケンシングに至るそれ以降のプロトコルのすべてに大きく影響することが、ここまででご理解いただけたのではないでしょうか。それでは、ソニケーターを準備して、クロマチンのシアリングを始めましょう!
PIXUL® Multi-Sample Sonicatorで先進のソニケーション
極めてばらつきの少ない断片化を提供し、最大96サンプルを並列処理可能でありながら迅速かつ低コストを実現した世界初そして唯一の多サンプルソニケーターPIXUL®についてもっと知るために、このショートムービーをご覧ください(日本語字幕選択可)。
こちらもご参照ください:
- PIXUL® Multi-Sample Sonicatorの詳細
- PIXULについてのお問い合わせ
- Nucleic Acids Research誌にて発表されたPIXUL-ChIPに関する記事
- Epigenetics PodcastにおけるPIXULのエピソード
- ChIP 101 eBookのダウンロード
- 最良のChIPデータを得るための手引き
- ChIPアッセイのエキスパートになる方法
About the author

Anne-Sophie Ay-Berthomieu, Ph.D.
Anne-Sophie was born in the south of France and grew up between the Mediterranean Sea and the Pyrenean Mountains. She grew up as a science fiction fan, leading her to specialize in molecular biology and genetics during graduate school at the University of Lyon, France (secretly hoping her research would give her superpowers!). After living in different places for work, she is back in Lyon, France where she shares her time between her husband, her family, and her friends. During her free time, Anne-Sophie challenges herself with hiking, climbing, racing, and traveling in foreign countries – while waiting for her superpowers to grow!
Contact Anne-Sophie on LinkedIn with any questions, or to tell her about your superpowers.
Related Articles
Guide to Generating the Best ChIP Data

March 15, 2019
The chromatin immunoprecipitation (ChIP) assay has become one of the most popular laboratory techniques to investigate the association of DNA-binding proteins and histones with chromatin. This article covers the major challenges of ChIP assays and how to overcome them to generate the best ChIP data.
Read More
Guide to Understanding the Benefits and Uses of Recombinant Antibodies
May 21, 2020
Antibodies are one of the most common, and most powerful, tools used by researchers. This article covers what recombinant antibodies are, and how they are different from polyclonal and monoclonal antibodies.
Read More
<< Back to MOTIFvations Blog Home Page



