<< Back to MOTIFvations Blog Home Page
ATAC-SeqやCUT&TagのライブラリーQCについて
「私のライブラリーは大丈夫?」
By Michelle Tetreault Carlson, Ph.D. and Marc Paradise
July 9, 2025
はじめに
ATAC-SeqやCUT&Tagは、クロマチン構造やDNA結合タンパク質を解析するための手法として、近年、多くの研究者に利用されるようになってきました。ATAC-Seqでは、アダプターが装填されたTn5を用いて、オープンクロマチンやヌクレオソーム、転写因子結合領域などの位置を検出することができます。一方、CUT&Tagでは、目的タンパク質に対する抗体と、アダプターが装填されたpA-Tn5を組み合わせることで、その結合部位付近のDNAを選択的に切断・タグ付けし、クロマチン上でのタンパク質の結合部位を同定します。どちらのプロトコルも、比較的簡単にシーケンシング可能なライブラリーを作製できることから、現在では広く用いられています。これらの手法の詳細については、「完全ガイド:ATAC-Seqの理解と使用法 」および「完全ガイド:CUT&Tagアッセイの理解と使用法」をご覧ください。
次世代シーケンシング (NGS) を行う前に、ほとんどの研究者はライブラリーの品質を事前に確認してシーケンスステップへ進みます。これは、NGS解析が高コストなため、期待されるデータ品質が得られないと判断された場合、解析へ進むことがコスト面で大きなリスクとなるためです。ライブラリー調製における最終的な品質確認 (QC) ステップでは、アダプターがライゲーションされたライブラリーのサイズ分布と濃度の両方をチェックしますが、ATAC-SeqやCUT&Tagを初めて実施する研究者にとっては、「どのようなサイズ分布の断片が理想的なのか」または「シーケンスに必要な量はどれくらいか」といった疑問を感じることも少なくありません。
厄介なことに、ATAC-SeqおよびCUT&Tagライブラリーのサイズ分布は、サンプルの種類や調製法によって大きく異なる場合があります。一般的なNGS解析用途として調製された通常のNGSライブラリーとは異なり、これらのライブラリーは、インタクトな核またはインタクトな細胞から直接調製されます。DNAのタグ付けには、核膜や細胞膜を通過して、内部のクロマチンにTn5トランスポゾームがアクセスすることが必要です。このアクセスは、細胞の種類、細胞数、細胞の健康状態、細胞の凝集状態、Tn5トランスポゾームとDNAの相対的な濃度によって影響を受けます。その結果、ライブラリー断片の量やサイズ分布に多くの多様性 (ばらつき) が生じる可能性があります。(Tn5トランスポゾームおよびpA-Tn5トランスポゾームについて、以降はトランスポゾームを省略し、Tn5、pA-Tn5と表記します。なお、全てアダプター配列を含むTn5酵素を意味します)
お客様から最も多く聞かれるのが、「このライブラリーのサイズ分布 (ライブラリートレース) は大丈夫そうですか?」という質問です。ライブラリートレースは、シーケンシングに進むかどうかを示す定性的な情報を与えてくれますが、ライブラリートレースを見ただけでシーケンシングが成功するかを判断することは容易ではありません。もちろん成功の可能性が高いパターン、あるいはライブラリー調製法に手を加えて再チャレンジした方が良いパターンなど、有用な情報を得ることはできます。本ブログでは、アクティブ・モティフの研究開発、受託解析サービス、テクニカルサポートの各チームが過去数年にわたり蓄積してきた、ライブラリーの解析方法に関するヒントやコツをご紹介していきます。
フラグメントサイズの解析
アクティブ・モティフでは、ATAC-SeqやCUT&Tagのライブラリーを評価する際に、ライブラリー増幅後のフラグメントサイズ分布の確認を推奨しています。その方法として、Agilent TapeStation (D1000 ScreenTapeアッセイ搭載) や、Agilent Bioanalyzer (DNA 1000チップ搭載) といったDNAフラグメントアナライザーの使用をお勧めしています。これらの機器を使うことで、フラグメントの電気泳動像を得ることができ、フラグメントのサイズとその分布を視覚的に確認することが可能です。なお、1000 bp以下の分解能を持つアッセイを選ぶことで、より正確なサイズ評価ができます。
CUT&Tagについて興味がありますか? CUT&Tagプロトコルの技術的な詳細や開発小話など、研究開発チームに所属するCasidee McDonoughとKyle Tanguayのポッドキャストを公開していますので、ぜひ聞いてみてください。
ライブラリーQC:ATAC-Seq
真核生物のクロマチンでは、核DNAがヌクレオソーム単位でパッケージされており、それぞれがヒストン8量体のコアに巻きついた147 bpのDNAを含み、20-90 bpの短いDNAリンカーによって各ヌクレオソームが繋がれています。このクロマチン内の遺伝子の密なパッケージングと緩いパッケージングにより、遺伝子の発現が制御されています。DNAがヒストンと緩く相互作用しているクロマチン領域 (オープンクロマチン領域) では、プロモーターやエンハンサーがDNAにアクセスできるようになり、転写が活性化されます。一方、DNAがヒストンと強固に結合しているクロマチン領域 (クローズドクロマチン領域) では、プロモーターやエンハンサー領域が隠されているため、転写を活性化することができません。
ATAC-Seqは、アダプターを装填したTn5がオープンクロマチンDNAに優先的にアクセスし、その部位のDNAを切断しつつアダプターを挿入するという特性を利用しています。これは、ヌクレオソーム間の20-90 bpリンカー領域内のどこででも起こり、リンカー領域内から90 bp未満の短い断片や、ヌクレオソームがTn5切断部に挟まれた大きな断片なども形成されます。これがATAC-SeqライブラリーのDNA断片のサイズ分布に反映されます。イルミナ社のP5/P7フローセルアダプターとインデックスを追加するライブラリー増幅反応により、ライブラリー断片には元のDNAインサートと、両端に付加されたアダプターの135 bpが追加されます。結果として、約200 bpから始まり、大きいものでは約1000 bp程度まで増加するライブラリー断片が形成され、隣接するヌクレオソームの周期性のため、断片は160-200 bp間隔のピークとして現れます。この周期的なパターンは、一般的にヌクレオソームラダー (nucleosome laddering) と呼ばれています。
サンプルの種類、細胞数、サンプルの取り扱いにばらつきがあるため、ライブラリーのサイズ分布 (大きさや形状) は異なることがありますが、おもに細胞の種類、健康状態、細胞数、Tn5とDNAの比率に依存します。サイズ分布におけるヌクレオソームのピーク数は、Tn5がDNAを切断できる頻度に依存し変動する可能性がありますが、その数や大きさと、良好なシーケンスデータとの間にはあまり相関関係がないことが分かっています。
ヌクレオソームラダー (nucleosomal laddering) は、高品質なDNAライブラリーであることを示す一つの指標となりますが、ヌクレオソームラダーを明瞭に得ようとする操作が、逆にライブラリー品質に悪影響を及ぼす場合もあります。
アクティブ・モティフのATAC-Seq kitは、オープンクロマチンのプロファイリングに特化して開発されたため、プロファイリングより高度な解析を目的とする場合には、別途、プロトコルの最適化が必要になる可能性があります。
一般的には、180〜600 bpの間に1〜3つのピークが見られ、800 bpを超える断片サイズが徐々に減少していくライブラリーは、NGS解析に適していると考えられます。最終的には、NGS解析の結果がアッセイの成功を判断する上で最も確かな指標となりますが、一方で、ライブラリーの断片化パターンは、アッセイ品質を評価するための初期的な目安として役立ちます。
40〜50 bp付近のピークは、プライマーダイマーによるものである可能性が高く、初期に使用する細胞数が少ない場合、プライマーが適切に結合するのに十分なDNAが得られず、プライマー同士が自己アニーリングしてダイマーを形成した可能性があります。また、少量のDNAに対してTn5のタグメンテーションを行う場合、短い断片や増幅可能な断片が少なくなるため、これもまたプライマーダイマーが生成されやすくなります。
SPRIビーズによるLeft-side selectionは、これらのダイマーを除去するために行いますが、最終的なライブラリーにはプライマーダイマーが少量残ることがあります。プライマーダイマーがライブラリー全体の5%未満であれば、SPRIビーズによる除去は必ずしも必要はないと考えていますが、ダイマーの割合が比較的高い場合には、追加のSPRIビーズによるLeft-side selectionを推奨します。
なお、ライブラリー全体の濃度がすでに低い場合は、実験を再実施するか、NGS解析に十分な量が残らない可能性があることを理解したうえで、SPRIビーズによるクリーンアップを行う必要があります。
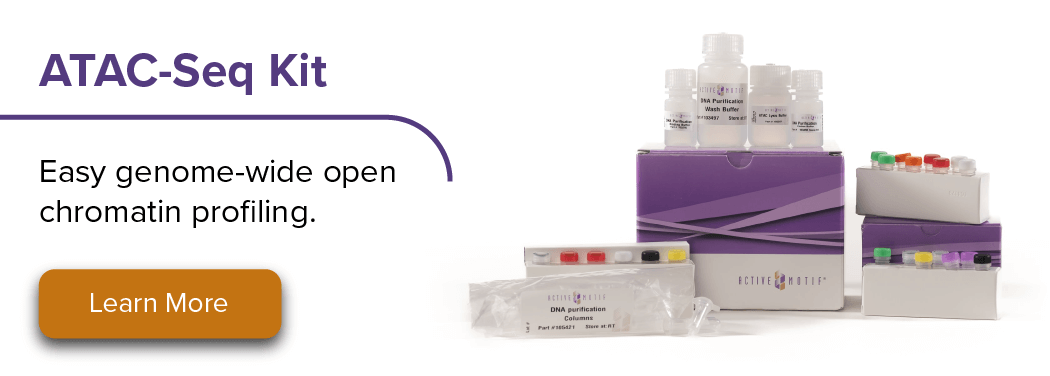
ATAC-Seqライブラリー フラグメンテーションプロファイル
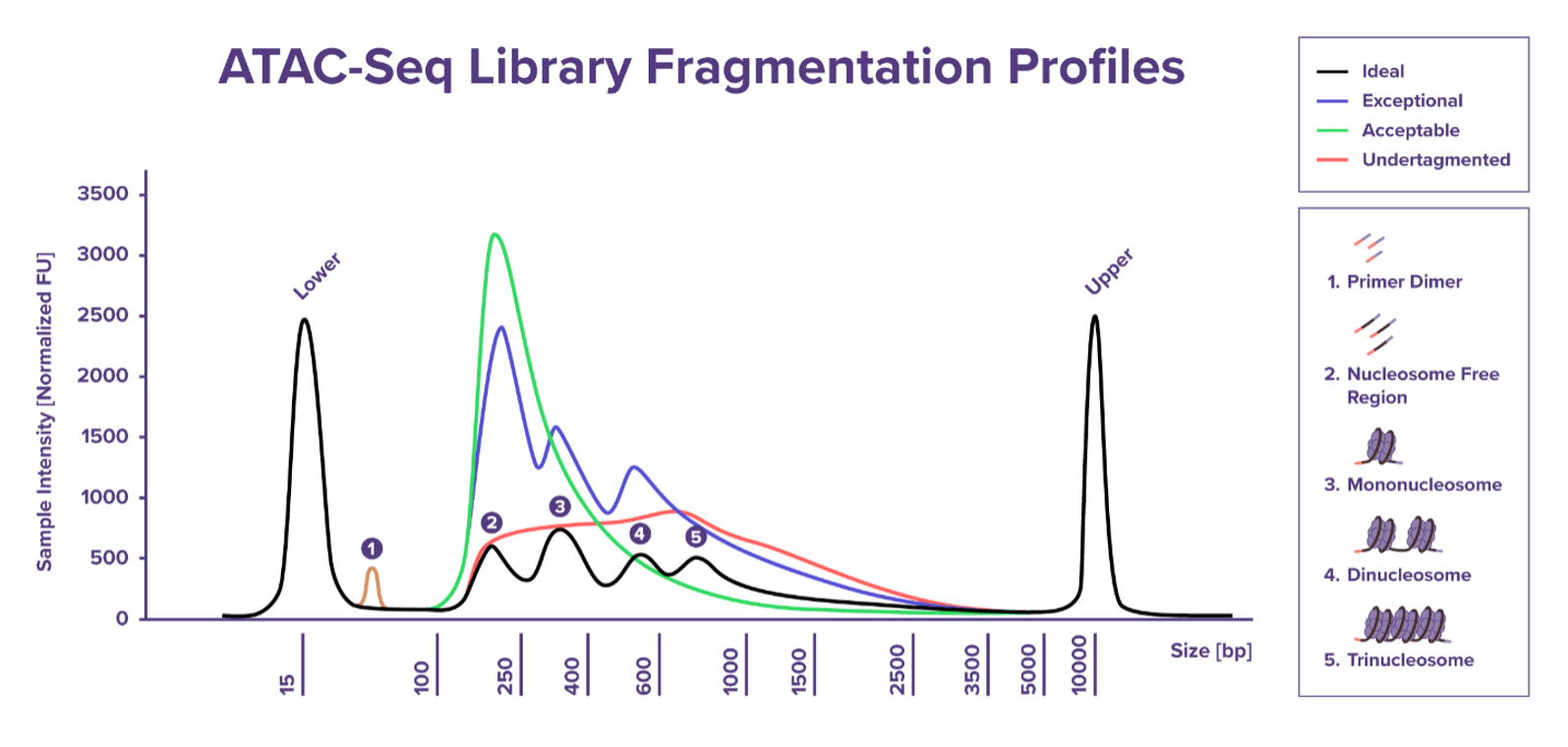
Fig.1 一般的なATAC-SeqライブラリーのDNA断片化パターンとピークについて
アンダータグメンテーション
Tn5が細胞内のオープンクロマチンに十分アクセスできない場合、あるいはTn5とDNAの比率が低すぎる場合には、DNAを十分な頻度で切断することができず、理想的なサイズ範囲の断片を生成することができません。このような状態はアンダータグメンテーションと呼ばれ、結果として得られる断片の多くが800 bpを超えるサイズとなってしまいます (Fig.2参照)。このような大きな断片はNGS解析においていくつか問題を生じます。まず、フローセル上でのクラスター化が非効率的となり、クラスター密度が低下するため、得られるシーケンスデータの総量が低くなります。さらに、小さなフラグメントに比べてシーケンス効率も劣るため、同じカバレッジを得るには、より多くのリードが必要となり、コストの増加につながります。
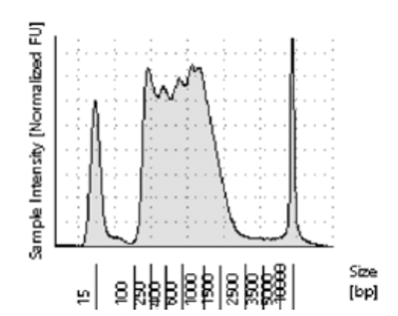
Fig.2 タグメンテーション不足によりほとんどの断片が800-2500 bpの間にあるATAC-Seqライブラリーの例
アンダータグメンテーション (under-tagmentation) が起こる原因はいくつかありますが、最も一般的なのは、核の調製が不十分なことによってTn5がクロマチンへアクセスしにくくなるケースです。特に、培養細胞に比べて組織サンプルでアンダータグメンテーションが起こりやすく、これは、核の分離や懸濁が困難で、組織を完全に単一細胞懸濁液するのが難しいため、細胞の溶解が不十分になるためです。
ライブラリーでアンダータグメンテーションが観察された場合は、以下を試してみてください:
- 溶解バッファーへの完全な懸濁を確実に行う (組織の場合はホモジナイズ処理を行う)。
- 氷上での溶解バッファー処理のインキュベーション時間を延ばす。
- タグメンテーション反応に使用するTn5の量を増やす、または使用する細胞数を減らす。いずれかの条件についてタイトレーションの実施を推奨します。
- サンプル中に溶解を阻害する物質が存在する可能性があるため、溶解前にPBSでの洗浄ステップを追加する。
もう一つの原因は、サンプルの品質が悪いことです。死細胞が多く含まれていると、アポトーシス由来のDNAが過剰に存在し、それがTn5の活性を飽和させてしまいます。その結果、クロマチンに結合したDNAへほとんど作用しなくなります。これを防ぐには、新鮮な細胞を使用するか、 50% FBS/40% 培養培地/10% DMSOなどの保存液を用いて適切な条件で凍結保存してください。組織を使用する場合は、切除後すぐに液体窒素でフラッシュフリーズし、-80℃で保存します。組織を扱う際は手早く作業を行い、氷上のペトリ皿に移して細かく刻むまで、決して組織断片を解凍しないように注意してください。
次に、Tn5と細胞数の比率についてです。活性をもつTn5酵素の量が多ければ多いほど、タグメンテーションがより多く行われ、その結果、DNA断片はより短くなります。当社のATAC-Seqキットのプロトコルは、5万~10万細胞で最適化されていますが、ライブラリーがアンダータグメンテーションを示し、他のすべての要因を検討しても原因が明確にならない場合は、タイトレーション試験が有効であると考えています。一例として、細胞数が2.5万、5万、10万、20万細胞になるように準備し、各反応でTn5の量は同じに保ち、サイズ分布を確認します。理想的な条件は、200~600bpの範囲に1~3つのピークが見られ、1000bpに近づくにつれて断片サイズが減少していくパターンです。また、使用できる細胞数が限られている場合は、タグメンテーション反応におけるTn5の量を段階的にタイトレーションすることも可能です。その際は、各反応で細胞数を一定に保ち、ddH₂Oの量を増減させて反応液全体の液量を一定にしてください。FACS (フローサイトメトリーによるセルソーティング) で分取された細胞は、ソーティング過程でダメージを受けることがあります。損傷を最小限に抑えるために、適切なソーティング技術を使用してください。また、可能な限り多くの細胞からスタートすることを推奨します。なお、標的細胞が10%未満の希少な集団である場合、高頻度 (例えば70%) の集団をソートする場合に比べて、標的細胞へのダメージが大きくなることに注意してください。
基本的な実験操作技術が、一貫性を保つうえで重要になります。1~2サンプルのみを調製する場合、一部の試薬のピペッティング量が0.5μLや2μLと非常に少量になることがあり、正確にピペット操作を行うことが難しくなります。可能であれば、タグメンテーション反応用のマスターミックスを作成し、複数のサンプルを同時に処理することで、サンプル間の一貫性を高めるようにしましょう。
ライブラリー調製の回復
タグメンテーションが不十分であったとしても、必ずしも最初から反応をやり直す必要はありません。SPRIビーズを用いたクリーンアップによって、大きな断片を除去することが可能です。アクティブ・モティフの標準的なATAC-Seqプロトコルでは、主に約150 bp以下のプライマーや小さな断片を除去する目的でSPRIビーズを使用しています。SPRIビーズを使ったクリーンアップでは、サンプルの一部が失われる可能性が常にあるため、通常は大きな断片を除去するステップを標準プロトコルに含んでいません。とはいえ、ライブラリーの断片の大部分が800 bp以上であることが確認された場合には、多少のサンプルロスがあっても、大きな断片を除去することでシーケンシングの効率が向上する可能性があります。そのため、そうした状況ではクリーンアップを実施する価値があると考えています。Fig.3には、DNA量に対して0.6倍のAMPure XPビーズ (SPRIビーズ) を使用してクリーンアップを行ったATAC-Seqライブラリーの例を示しています。クリーンアップ後は、全体のDNA量が減少しているものの、NGS解析に必要な量は十分に残っていることがわかります。ライブラリーの収量に関する詳細は、次の項目をご参照ください。
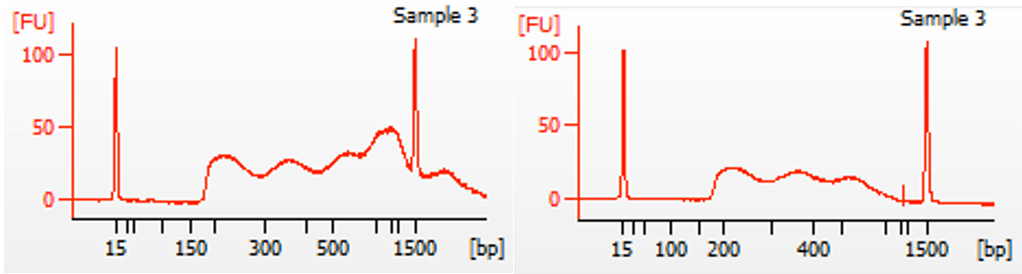
Fig.3 AMPure XPビーズをDNA量の0.6倍量用いたライトサイドセレクションによるDNAクリーンアップ (左:クリーンアップ前、右:クリーンアップ後)
ライブラリー収量:ATAC-Seq
よくある質問のひとつに、「ATAC-Seqの調製でどれくらいのライブラリー収量が得られますか?」という問い合わせがあります。理想的な条件下、たとえば、5万~10万個の新鮮な細胞を用いて、プロトコルを完全に実行したとすると、400~600 ngのライブラリーが得られることがあります。Active Motifのキットでは、これが20 µlの溶出バッファーに溶出されるため、濃度としては約20~30 ng/µlになります。一方、FACSで分取した細胞では、同じ細胞数でも細胞ダメージの影響により収量が低くなる傾向があります。また、そもそも5万個の細胞が用意できない場合には、それに応じて得られるライブラリー量も少なくなります。さらにSPRIビーズによるクリーンアップの各ステップでは、試料をロスしやすいために、得られるDNA量が少なくなることがあります。試料のロスを最小限に抑えるためには、エタノールをチューブの底にそっと加え、磁気ビーズのペレットを乱さずにゆっくりと上昇させるようにします。また、磁気ビーズを過度に乾燥させないよう注意しましょう。ビーズがひび割れると、試料損失の原因になります。そして、DNAのカラム精製を行う際は、溶出バッファーをカラムの中央に丁寧に添加するようにしてください。
幸いなことに、NGS解析では少量のDNAしか必要ありません。Active Motifの受託解析サービスでは、時には20 ng (1 ng/µl) ほどのサンプルでも良好なデータでNGS解析が行われています。もしライブラリーをプールしてシーケンサーにかけられるだけのDNA量があり、断片の状態も良好であれば、NGS解析に進む価値は十分にあると考えられます。
ATAC-Seqのコツ:
- 200〜600 bpの範囲で1〜3本のピークが見られるのが理想的で、800 bpを超える断片は徐々に減少しているか確認してください。
- 最終的なライブラリー濃度は20〜30 ng/µlが望ましく、1 ng/µl以上であれば許容範囲です。
- プライマーダイマーが5%を超える場合は、SPRIビーズによる2回目のクリーンアップで除去してください。
- 断片化が不十分で大きな断片 (600 bp以上) が多く含まれる場合、200〜600 bp範囲に十分なライブラリーがあれば、SPRIビーズ精製によるRight-side selectionでそれらを除去できます。
- 細胞品質の次に、Tn5と細胞数の比率が断片化パターンに最も大きな影響を与えます。サンプルの種類に応じてこの比率を最適化する必要があります。
- PCRサイクル数を増やしても、データ品質の向上には限界があり、細胞数・細胞品質・適切なTn5と細胞の比が重要です。
ライブラリーQC:CUT&Tag / CUT&Tag R-loop
CUT&Tagプロトコルでは、タグメンテーションの前に核を単離するための細胞溶解ステップなどはありません。まず、生きた細胞を磁気性のコンカナバリンAビーズに結合させ、その後、ヒストン修飾や一部の転写因子などDNA上の標的に対して一次抗体を結合させます。次に、シグナル増強のために二次抗体を結合させ、そこにアダプターを装填したpA-Tn5を導入し、二次抗体が結合している位置でDNAを切断します。ATAC-Seqとは異なり、CUT&TagではpA-Tn5が抗体の結合部位でのみ切断を行うため、小さな断片はそれほど多く生成されません。電気泳動によるライブラリートレースでは、隣接するヌクレオソーム間の距離に由来するモノヌクレオソームおよびオリゴヌクレオソームのラダー構造が見られます。DNA上のアダプターの長さが135 bpで、隣接するヌクレオソーム間のピーク間距離が150〜200 bpであるため、ヌクレオソームラダーはおおよそ150〜200 bp間隔で現れます。約350 bpの位置に見られるピークはモノヌクレオソーム、約550 bpの位置に見られるピークはダイヌクレオソームに対応します。はっきりとしたモノヌクレオソームのピークが理想的とされますが、サンプルの種類や条件によってパターンは変動するため、200〜600 bpの範囲内にピークが見られることを重要と考えてください。Fig.4では、 K562細胞を用いて、H3K27me3を標的とした際のライブラリートレース例を示しています。
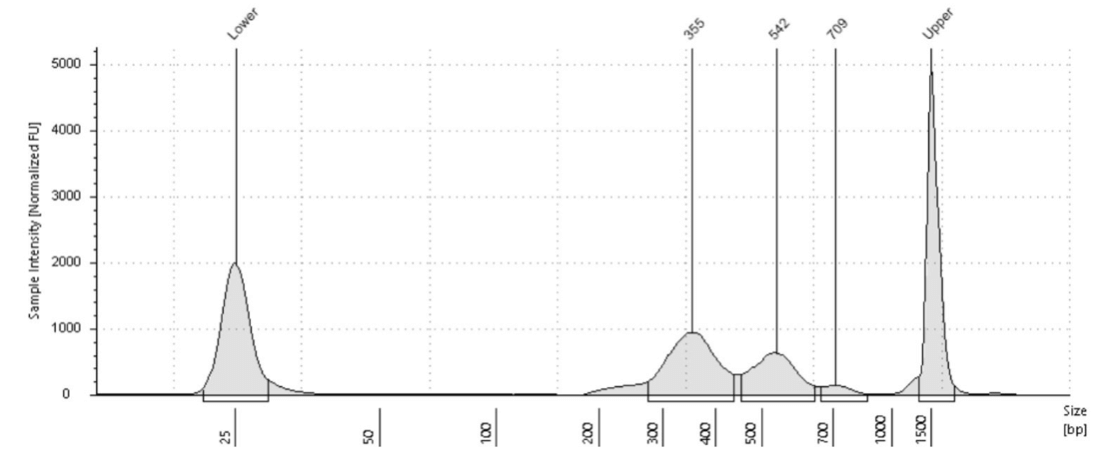
Fig.4 H3K27me3抗体を用いたK562細胞10万個からのCUT&Tagライブラリーの例
抗体やpA-Tn5と細胞数との比率にわずかな違いがあるだけでも、ライブラリートレースにおけるピークの高さが変動することがあります。しかし、Kay-Okur et al, (2019)の論文で示されているように、こうした変動は重大な問題ではなく、シーケンシングデータとしては一貫性のある結果が得られることが分かっています。Fig.5では、タグメンテーション反応に加えるpA-Tn5の量を変化させた場合に、ライブラリーのピークにどのような違いが生じるかを示しています。pA-Tn5の濃度が低い条件では、大きなピークがより高いフラグメントサイズへとシフトしています。それでも、これらすべての条件において、正規化されたシーケンストラック (カバレッジプロファイル) は同一であったことが確認されています。
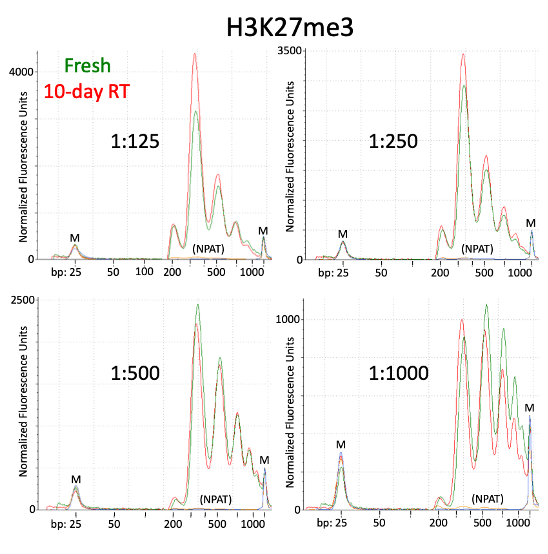
Fig.5 Kay-Okur et al, (2019)より引用した図は、CUT&Tag によって作製されたライブラリーのトレースデータを示しています。対象は、ヒストン修飾である H3K27me3 です。タグメンテーションの際には、pA-Tn5 の希釈倍率を変えて使用しました。反応後、1.1倍のAMPureビーズでクリーンアップを1回行い、DNAは 10 mM Tris-HCl (pH 8) 25 μl で溶出しました。その後、2 μlのサンプルを用いて TapeStation D1000 による解析を実施しました。マーカー (M) :Lower Marker = 25 bp、Upper Marker = 1500 bp。
ライブラリー収量:CUT&Tag
転写因子のように発現量が少ない標的に対する抗体を使用した場合や、細胞数が少ない (2万個未満) 場合、電気泳動像上でライブラリーがほとんど、またはまったく見えないことがあります。 このような状況では、NGS解析に進むべきかどうかの判断が難しくなります。こうしたケースでは、プロトコルが正常に機能しているかを確認するために、ポジティブコントロールを同時に実施することが有効です。CUT&Tag法の開発者であるSteven Henikoff 博士が指摘しているように、H3K27me3由来のフラグメントは検出しやすく、その理由は、多くの細胞種においてゲノム全体の約3% (ヒトゲノムで約1億塩基対) をカバーしているためです。一方、結合部位が約10,000箇所しかない転写因子の場合、ゲノムカバレッジはおよそ100万塩基対にとどまり、ライブラリーフラグメント解析で検出できない可能性があります。アクティブ・モティフの受託解析サービスでは、H3K27me3のようなポジティブコントロール抗体で良好なシグナルが確認できた場合、たとえ別の抗体ではライブラリーが視認できなかったとしても、ライブラリーの定量へ進める価値があると考えています。目安として、ライブラリー濃度が少なくとも1 ng/μl (平均360 bp換算で約4 nM) 以上ある場合は、シーケンスに進むことを推奨しています。Fig.6では、K562細胞およびH3K27ac抗体を使用し、非常に少ない量のライブラリー例を示しています。このライブラリーでも、最終的にはNGS解析に十分な量のDNAが得られました。
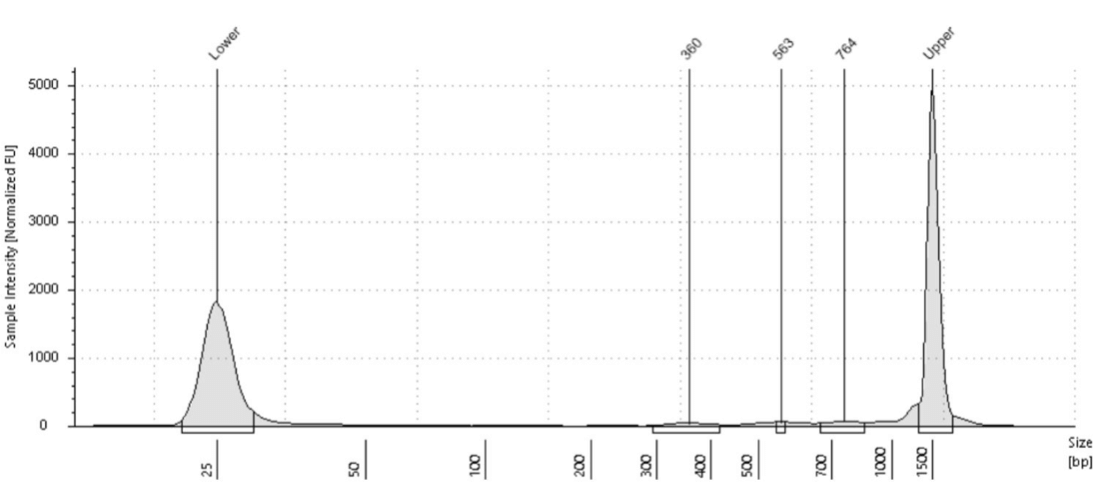
Fig.6 H3K27ac抗体を用いたK562細胞10万個からのCUT&Tagライブラリーの例 収量はすくないが、シーケンシングに成功し、高品質な解析データを得るために十分なDNAが得られています。
ライブラリーの定量:一般的なガイドライン
NGS解析用にライブラリーをプールする際は、フローセル上でクラスタ密度が最適化され、すべてのライブラリーでリード数が均一に分布するように、ライブラリーの濃度を正確に調整することが重要です。最適な濃度は、使用するシーケンサーやシーケンス試薬キットによって決まります。複数のライブラリーを混合すると、それぞれのライブラリーは他のライブラリーによって希釈されるため、シーケンサーに投入する最終的なライブラリー濃度よりも、個々のライブラリーの最低必要濃度は高く設定する必要があります。
Illumina装置でのNGS解析用ライブラリーの定量には、KAPAライブラリー定量キットのようなqPCRベースのアッセイを推奨します。TapeStationやBioanalyzerで収量を推定することもできますが、正確さは劣ります。KAPAライブラリー定量は、Illuminaフローセルに結合するP5およびP7アダプター配列を含むライブラリー断片のみを選択的に増幅することで機能します。このため、理論的にはNGS解析が可能なDNAだけが定量されます。NanoDropのような分光光度法やBioanalyzerのような電気泳動法は、アダプターの有無にかかわらずすべてのDNAを定量してしまうため、KAPA法のほうがより正確な定量性をもちます。
qPCRベースのアッセイでは、増幅された各ライブラリの定量サイクル数 (Cq) をDNA標準品のCq値と比較します。そこから標準曲線を用いてライブラリー濃度を算出します。モル濃度はDNA断片のサイズに依存します。単一で狭く均一なピークを持つ多くのライブラリー調製ではこの方法は簡単ですが、ATAC-SeqやCUT&Tagライブラリーは断片サイズの分布が広く、複数のピークを持つことが多いため、実効濃度の決定はやや複雑になり、多くの断片が存在する領域や、最も高いピークの位置を確認して、それを基準にする必要があります。モル濃度から二本鎖DNAライブラリー濃度を計算したり、ライブラリーをプールする方法については、Illuminaのウェブサイトでサポート情報が提供されています。
トラブルシューティング
H3K27me3のようなポジティブコントロール抗体でライブラリーが作製できるのに、他の標的で失敗する場合、それはCUT&Tagに対して検証されていない抗体を使用していることが原因かもしれません。また、転写因子を標的にしている場合は、CUT&RunやChIP-seqといったアッセイの方が適している可能性があります。
検証済みの抗体を使用していてもライブラリーがほとんど、または全く得られない場合、通常はサンプルの品質やサンプルの損失に問題があることを示しています。まず、核染色 (例:トリパンブルーやAOPI) によって生存が確認された細胞から開始することが重要です。細胞の生存率は70%以上が推奨されます。単離した核を使用する場合は、高品質な核を使用する必要があり、核のブレッビングや断片化がほとんど、あるいは全く見られない状態であることが理想です。組織から始める場合は、カウントされた細胞や核の数が、破片などが誤って含んでいないか確認してください。この目的にはAOPIの方が一般的に優れています。詳しくは、当社のセルカウント技術ノートをご覧ください。
アッセイを始める前に、コンカナバリンAビーズ (Con Aビーズ) が凝集していないことを確認してください。Con Aビーズは凍結温度に非常に敏感です。もし凝集が確認された場合は、そのビーズは使用せず、Active Motifのテクニカルサービスに連絡してください。凝集の確認には、Con Aビーズを5〜10 µl取り、同量のPBSで希釈し、ヘモサイトメーターまたはカバーガラス付きスライドガラスを用いて顕微鏡で観察します。均一に分散した小さく淡い茶色の粒子であれば使用可能です。一方、大きく濃い茶色の塊が見られる場合は凝集しているため、ビーズの交換が必要です (詳細は使用しているプロトコルを参照してください)。
Con Aビーズの凝集が原因でない場合、次に確認すべきは細胞がCon Aビーズに正常に結合しているかどうかです。これは、細胞とビーズを混合し、マグネットスタンドに置いたステップの後に確認できます (詳細は使用しているプロトコルを参照してください)。上清を破棄する前に、バッファーの10 µlをヘモサイトメーターで観察します。正常に結合していれば、上清中に細胞はほとんど、あるいは全く存在しないはずです。正常なビーズであれば、未結合の細胞数は全体の5%未満であることが一つの目安となります。それでも細胞がビーズに結合しない場合、原因は培養細胞の剥離や組織の解離時にトリプシンのような消化力の強い酵素を使用した可能性が考えられます。細胞膜上の糖タンパク質はCon Aビーズとの結合に必要であり、それを損なわないTrypLEやAccutaseといった試薬の使用を推奨します。
もう一つ重要な検証事項は、ジギトニンを含む抗体バッファーを添加した後に、トリパンブルーを用いて細胞の透過処理が成功しているかを確認することです。ジギトニンは非常に重要な成分であるため、まずは抗体バッファーにジギトニンが確実に添加されているかを確認してください。次に、ヘモサイトメーターとトリパンブルーを用いて、すべての細胞が青く染色されていることを確認します。この問題はあまり一般的ではありませんが、特にリンパ球のように透過処理が困難で知られる初代細胞を扱う場合には、確認が重要です。細胞が透過されていない場合は、ジギトニンが添加されていない、あるいは濃度が不足している可能性があります。
CUT&Tag R-loop
Active Motif製のCUT&Tag-IT® R-loop Assay Kit は、 CUT&Tagプロトコルをベースに、 R-loopのプロファイリングに最適化された製品です。キットに含まれるRNase Aは、サンプル中のR-loopを分解して除去するため、ネガティブコントロールとして機能します。当社の検証では、RNase Aで処理したサンプルが、未処理のものと比較してライブラリー収量が5〜35倍低下することを確認しています。
文献では、 R-loopの分解に最適な酵素としてRNase Hがよく挙げられていますが、検証の結果、RNase Hは細胞内でRNA:DNAハイブリッドを効率的に切断できないことがわかり、これに対してRNase Aがこのアプリケーションにおいてより効果的であることを確認しました。
他のCUT&Tagシリーズのプロトコルと比べ、いくつか違いはあるものの、ライブラリーの一般的な特性やトラブルシューティングは殆ど同じです。このアッセイから得られるライブラリートレースは、通常200〜600 bpの範囲に1〜3本のピークを示し、全体的なライブラリー収量は、豊富なヒストン修飾マークを対象とした場合と比べてほとんど低くなります。未処理サンプルに対してRNase A処理サンプルのライブラリー収量が5〜35倍低い場合には、収量が非常に少ないライブラリーでもNGS解析を行う価値があります。推奨されるトラブルシューティング手順を実施してもライブラリーの増幅が確認できない場合は、H3K27me3 (Cat# 39156) のようなポジティブコントロール抗体の使用を検討してください。これにより、アッセイが正しく実施されているかどうかを判断することができます。
なお、ある種の生物学的サンプルではR-loopの含有量が非常に低く、検出が困難な場合があります。そのような場合は、 R-loopが豊富に存在することが知られているHeLa、HCT116、またはK562といったポジティブコントロール細胞株を用いて、問題がサンプルの種類によるものかどうかを確認することをお勧めします。
Note that some biological samples may contain very low levels of R-loops, making detection difficult. In such cases, it is advisable to test a positive control cell line known to contain abundant R-loops, such as HeLa, HCT116, or K562, to determine whether the issue lies with the sample type.
CUT&Tag-IT® R-loop ライブラリー例
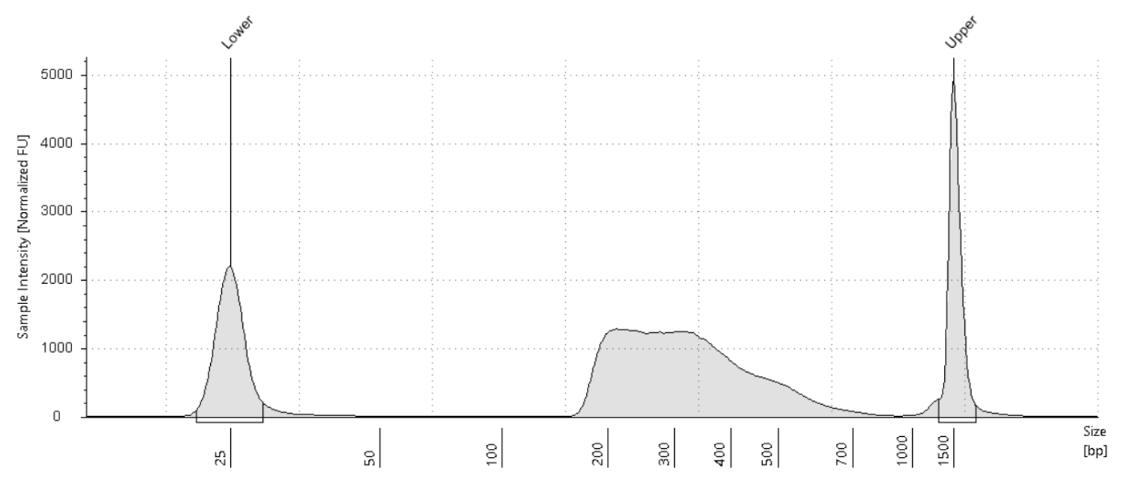
Fig.7 K562細胞50万個のライブラリー
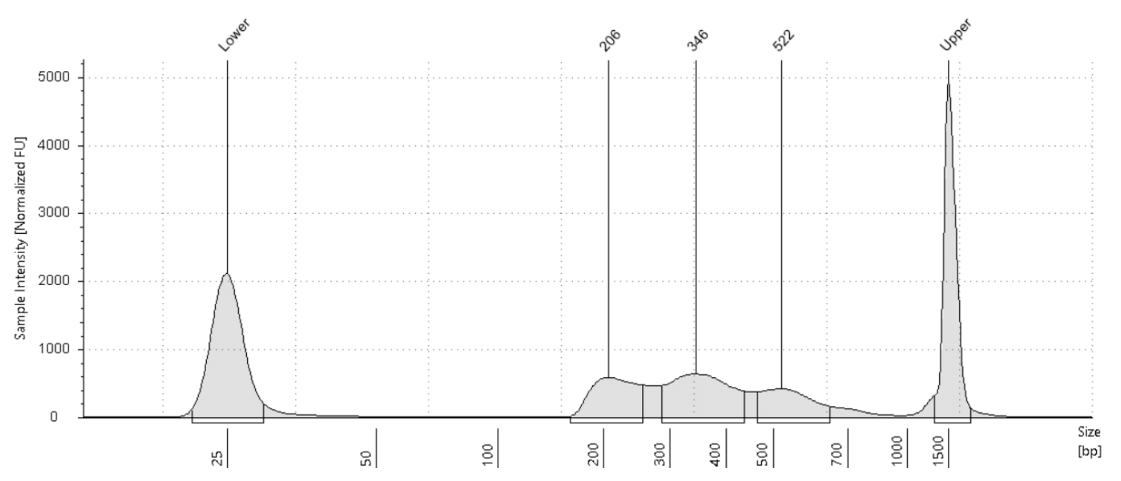
Fig.8 Jurkat細胞50万個のライブラリー
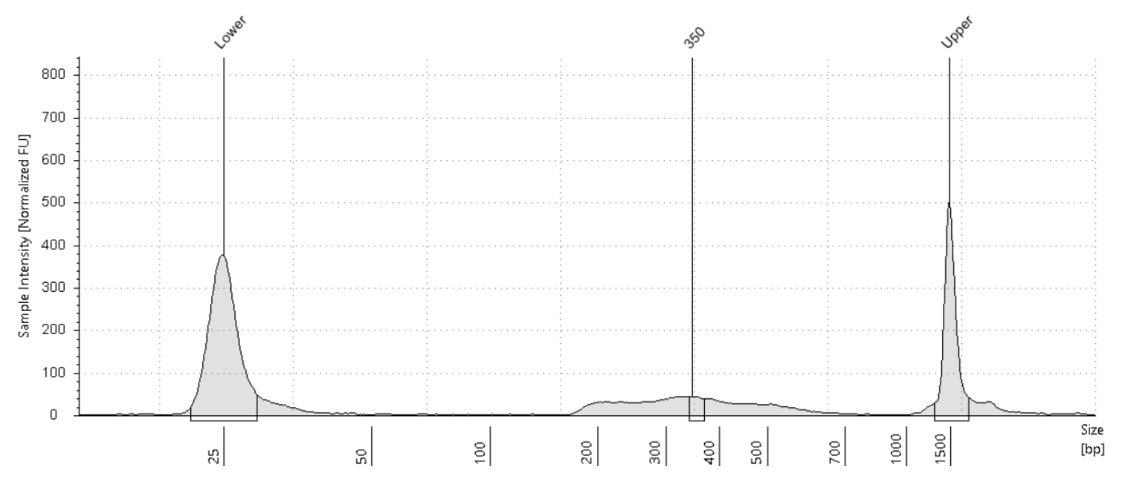
Fig.9 RNase Aで処理したネガティブコントロール
CUT&Tagのコツ:
- 200〜600 bpの間に単一のピークがあり、全体の濃度が1 ng/µl以上のライブラリーの場合、NGS解析へ進むことを推奨します。約350 bp付近に強いモノヌクレオソームピークが見られるのが理想的とされていますが、必須ではありません。
- ポジティブコントロールの収量が十分であり、NGS解析に必要な量のライブラリーが確保できているようならば、より収量の低いライブラリーからでも有用なデータを得られる可能性があります。
- ライブラリーが確認できない場合は、以下の要因を検討してください:
- 細胞数
- 細胞の品質/生存率
- ConAビーズの状態
- ConAビーズの結合容量
- 細胞の透過効率

まとめ
ATAC-SeqやCUT&Tagでは、ライブラリーフラグメントのサイズ分布にばらつきが生じることがあります。同じ試薬を毎回安定して含むキットを使用していても、細胞数、細胞の溶解状態、特定のヒストンマークの頻度など、わずかな違いによってTn5やpA-Tn5の切断頻度に差が生じ、それが断片化に影響を与えることがあります。しかし、このようなばらつきがあっても、Tn5がオープンクロマチンにアクセスできるか、あるいはpA-Tn5が抗体に結合できるかに依存するため、シーケンスデータの品質自体は安定して良好な場合が多いです。そのため、ライブラリーのサイズ分布は、シーケンスを進めるべきかどうかを判断するための定性的な指標として捉えてください。
ライブラリーの定量は、QubitやqPCRベースのアッセイを用いることで、より正確に行うことができ、フローセル上での効率的かつバランスの取れたNGS解析にもつながります。
上記のガイドラインが、ライブラリーを次のステップへ進めるかどうかの判断に役立てば幸いです。ご不明な点がございましたら、Active Motifのテクニカルサポート ([email protected]) までお気軽にお問い合わせください。
References:
https://www.sciencedirect.com/science/article/pii/S2666166720300666
Holly Brunton, Ian M. Garner, Ulla-Maja Bailey, Rosie Upstill-Goddard, Peter J. Bailey, Using Chromatin Accessibility to Delineate Therapeutic Subtypes in Pancreatic Cancer Paient-Derived Cell Lines, STAR Protocols, Volume 1, Issue 2, 2020,100079, ISSN 2666-1667 https://doi.org/10.1016/j.xpro.2020.100079.
https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4559-3
Ou, J., Liu, H., Yu, J. et al. ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-Seq data. BMC Genomics 19, 169 (2018).
https://doi.org/10.1186/s12864-018-4559-3.
https://www.nature.com/articles/s41467-019-09982-5
Kaya-Okur, H.S., Wu, S.J., Codomo, C.A. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019).
https://doi.org/10.1038/s41467-019-09982-5 .
https://www.protocols.io/view/bench-top-cut-amp-tag-bcuhiwt6?step=66
A special thank you to scientists Jesse Lopez and Casidee McDonough in Active Motif Services for their technical expertise on ATAC-Seq and CUT&Tag.
About the authors

Michelle Tetreault Carlson, Ph.D.
Michelle’s interest in science was first spurred by the starry skies above her rural farm in upstate New York State, leading her to pursue a B.S. in physics. She was originally interested in astrophysics when entering the University of California, San Diego, but transitioned towards the more practical pursuit of biology earning her Ph.D. in Biophysics, studying photosynthetic proteins. Michelle’s postdoctoral research on retinal ion channels, took her further towards biology, ultimately leading to a career in the biotech industry. She enjoys chatting with scientists about their projects and interacts with them both as a Technical Support Scientist and Product Manager for Active Motif’s DNA Methylation products.
Michelle is a mother of 4 kids and 2 cats, and her hobbies include puzzles (the sign of a patient and logical mind), cooking, and pondering the human condition.

Marc Paradise
Marc grew up in New York and served as a U.S. Navy rescue swimmer and aircrewman aboard SH-60 helicopters before beginning his academic career. He earned his B.S. in biochemistry and cell biology from the University of California, San Diego. Marc went on to manage a research lab at the UCSD Moore’s Cancer Center, where he focused on cancer immunology—particularly macrophage phenotypes in the tumor microenvironment— he also established and ran a 10x Genomics single-cell sequencing core for the center. He is currently a Technical Support Scientist at Active Motif, helping researchers advance their work in epigenetics. Outside of the lab, Marc enjoys golf, as well as strength and conditioning sports.
Related Articles
Comprehensive Guide to Understanding and Using CUT&Tag Assays

November 24, 2020
CUT&Tag shows a lot of promise and has the potential to alleviate some ChIP limitations, but it also has its own set of limitations that must be considered. This article covers what CUT&Tag is and describes the advantages and drawbacks of this method.
Read More
Complete Guide to Understanding and Using ATAC‑Seq

February 9, 2021
The ATAC-Seq method was just published in 2013 and it has already become one of the most common and powerful ways to study chromatin states on a genome-wide level. This article covers what ATAC-Seq is and how to use it in your research.
Read More
<< Back to MOTIFvations Blog Home Page



