<< Back to MOTIFvations Blog Home Page
Guide to Understanding and Using Hi-C and Related Chromosome Conformation Capture Assays

April 20, 2020
DNA was first visualized in the mid-nineteenth century; an observation that was only possible for DNA in its most condensed form – which generated the now-familiar X-shape we know as the chromosome. Fast forward roughly 160 years to the completion of The Human Genome Project, and the realization that the linear genomic sequence alone would not be enough to unlock the secrets of gene regulation.
Since then, we have broadened our understanding of the human genome to include its dynamic nature and complex 3-dimensional structure, and the hundreds of thousands of proximal and long-range interactions that contribute to gene regulation within a chromosome.
Research on intra-chromosomal interactions was limited by inadequate techniques until 2002, when the lab of Job Dekker published the first biochemical approach for observing long-range chromatin interactions which he called chromosome conformation capture, or 3C for short. While this method proved invaluable for understanding chromatin contacts at a small scale, it wasn’t until the advent of next-generation sequencing (NGS) technologies that the era of 3-dimensional chromatin took hold.
Advancement in high-throughput DNA sequencing technologies has allowed the focus to shift towards a more thorough mapping of chromatin architecture. In line with that trend, the original 3C method has been successively upgraded, with each iteration incorporating new and improved technologies. This has produced a great variety of chromosome capture techniques such as 3C, 4C, 5C, Hi-C, ChIP-loop, ChIA-PET, TCC and T2C, HiChIP, PLAC-seq, and more!
While the acronyms may be numerous, these varying techniques ultimately all aim to address components of the same chromosome structure question.
Learn More About Active Motif's End-to-End Hi-C Service
What is Chromosome Conformation Capture?
Chromosome conformation capture is a research method that allows researchers to observe interactions between genetic loci that are in close contact in the 3-dimensional structure of a chromosome but can be megabases apart in the linear sequence.
These long-distance relationships are difficult to predict based on linear sequence alone, but can heavily influence gene expression. These methods also provide information on the 3-dimensional organization of the genome in the nucleus. Spatial organization of the genome has been implicated in playing a role in the regulation of many biological processes including transcriptional regulation, differentiation, gene silencing, cell cycle regulation, and disease.
All of the chromosome conformation capture assays (and their derivatives) share the same basic principles and to some degree, have overlapping methodologies. The unifying principle across these various methods is the “freezing” of DNA-DNA interactions via chemical crosslinking. The DNA is then digested, and the crosslinked fragments are ligated together, creating a hybrid DNA sequence composed of the two loci that were interacting. From here, the methods diverge in terms of isolating and identifying these fragments, but ultimately, they are sequenced and mapped back onto the genome to determine where these interactions occur.
A Brief History of 3C, 4C, 5C, and Similar Methods
We have known for a long time that the DNA present in chromatin within the nucleus is not linear, but until relatively recently it has been difficult to investigate the 3-D conformation of chromatin.
There are now many different ways to investigate the 3-D structure of chromatin. In fact, the sheer number of available chromosome conformation capture methods can be overwhelming at first glance. Below we discuss the basic methodology and applications for the most commonly used subset of chromosome conformation capture-based assays.
Chromosome Conformation Capture (3C)
Chromosome conformation capture (3C) was the first chromatin structure assay on the scene in 2002. This method is the foundation on which many subsequent technologies were built – they improve upon this method in some ways but still follow the same general workflow.
In this original method, the cells are first treated with formaldehyde to crosslink interacting loci. Cells are then lysed, and the DNA is subjected to restriction digestion to generate small fragments containing the cross-linked DNA strands. The DNA sample is then massively diluted so that when the subsequent ligation step is performed, it favors ligation of strands within a crosslinked pair, rather than between non-cross-linked strands. The crosslinking is then reversed, and the samples undergo qPCR amplification and detection. From this, the relative frequency of each contact (representing two interacting loci) can be calculated.
The qPCR primers are designed to amplify the loci of interest, which requires prior knowledge of the genes that might be interacting and therefore limits 3C to observing a singular interacting pair of loci. For this reason, it is often referred to as a “one-vs-one” approach and is primarily used for validating candidate interactions such as long-range promoter-enhancer pairs.
Chromosome Conformation Capture-on-Chip (4C)
The first upgrade to 3C was in the form of chromosome conformation capture-on-chip (4C), which was published in 2006.
The workflow for 4C mimics that of 3C up until the crosslinking is reversed. From here, the hybrid strand is digested again to create 5’ overhangs, which are then ligated to produce circularized DNA fragments. PCR primers are designed to sit down at a known, chosen locus of interest and amplify outward. The amplified library is then analyzed using a microarray chip.
4C reveals interactions between one known locus and all other genomic regions (one-vs-all). 4C is particularly useful for identifying novel interactions or changes to a singular locus; for example, investigating a gene involved in development or disease.
Chromosome Conformation Capture Carbon Copy (5C)
After 3C and 4C, the logical progression was to find a way to measure many different interacting regions at once (many-vs-many). Thus, chromosome conformation capture carbon copy (5C) entered the scene in 2006.
In 5C, a region of interest (typically around one megabase) is queried for all interacting loci within it. 5C also follows the same initial steps as 3C and 4C, with crosslinking, digestion, ligation, and reversal of the crosslinking performed in the same fashion. Now diverging from the earlier methods, the sample library undergoes ligation-mediated amplification (LMA). In LMA, primers are annealed to the sequences created by the restriction digestion, which, after the ligation step, now reside in the center of the hybrid strand. The primers are annealed, creating a hybrid DNA strand with two primers at its center. Only strands containing the dual primer set can be amplified, creating a “carbon copy” of the originally interacting loci (the 5C library).
5C requires no prior knowledge of the loci to be assayed, thus overcoming a major limitation of 3C and 4C. And while 5C does allow for the observation of multiple interacting loci within a region, it is still relatively low-throughput and unsuitable for genome-wide interrogation.
Hi-C
These early 3C-based methods culminated in the development of Hi-C in 2009, providing an “all-vs-all” approach for genome-wide identification of chromatin interactions.
As with its predecessors, Hi-C begins with the common cross-linking and DNA digestion steps. But post-digestion the 5’ overhangs are filled in, incorporating biotinylated nucleotides. After blunt-end ligation and reversal of the cross-links, the biotin tag is at the center of the ligation junction in the chimeric DNA strand. Fragments containing the internal biotin tag are pulled down using streptavidin beads, purifying the sample such that it is primarily composed of hybrid strands representing informative contacts. The samples undergo next-generation sequencing (NGS) and are mapped back onto the genome to determine the frequency of the observed interactions.
Hi-C was simultaneously the first genome-wide and the first unbiased approach to investigate chromatin conformation, as it required no choice of locus or region as a viewpoint.
Learn More About Active Motif's End-to-End Hi-C Service
Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET)
A second genome-wide technique also debuted in 2009 called chromatin interaction analysis by paired-end tag sequencing (or ChIA-PET for short). ChIA-PET combines chromatin capture with chromatin immunoprecipitation and paired-end tag sequencing to concentrate and analyze long-range chromatin interactions.
In this method, an antibody against a protein of interest is used to pull-down the protein and any DNA cross-linked in the complex (chromatin immunoprecipitation). The samples are sonicated to fragment the DNA, and biotinylated linkers are added prior to ligation. The biotinylated strands are pulled-down using streptavidin beads, and the samples are mapped using high-throughput paired-end tag sequencing.
ChIA-PET excels in protein-focused research questions and has routinely been used for mapping the interactions of various transcription factors.
Tethered Conformation Capture (TCC)
With Hi-C finally solving the genome-wide piece of the puzzle, further endeavors aimed to refine these chromosome capture techniques. Tethered conformation capture (TCC) was born from this effort in 2011 and improves upon Hi-C by performing the ligation step on a solid substrate rather than in solution.
In TCC, both the proteins and the DNA fragments are biotinylated, and the biotinylated proteins are immobilized on a solid surface for ligation, which reduces the occurrence of random ligation and increases the specificity of the assay. After the cross-linking is reversed, the biotinylated chromatin is pulled down, providing a second purification/concentration step. As in ChIA-PET, the fragments are amplified by paired-end tag sequencing.
Targeted Chromatin Capture (T2C)
Targeted chromatin capture (T2C) was developed in 2014, but it takes a step back and improves upon the “many-vs-many” approach used in 4C. T2C provides a more in-depth, higher resolution view of a particular region of interest, assaying the interactions between all loci within.
The workflow is similar to that of 4C: cross-linking and restriction digest followed by ligation and a second digest. Next, T2C borrows from other capture methods, using adaptor ligation and pull-down to immobilize the strands on beads or an array, and the fragments are sequenced using the paired-end tag.
T2C provides a more cost-effective alternative to Hi-C, provided there is a specific and known locus to sample rather than the need for a genome-wide viewpoint.
HiChIP & PLAC-Seq
The most recent additions to this list are HiChIP and PLAC-seq, both of which were published in late 2016. Like ChIA-PET, these methods are more protein-centric views of the chromatin landscape, but work with smaller sample sizes or fewer cells, without compromising resolution.
To achieve high-quality results with fewer cells, HiChIP carries out the crosslinking, biotinylation, and ligation steps all within the nucleus, prior to cell lysis. This confined space ligation step decreases the number of false-positives and improves the efficiency of the ensuing chromatin immunoprecipitation step. Paired-end sequencing is used to identify fragments and interactions, which can be mapped back onto the genome.
PLAC-seq also takes advantage of the confines of the nucleus for increasing efficiency, performing the cross-linking, restriction digest, biotinylation, and ligation in the intact nucleus. And unlike traditional ChIA-PET, the ligation step occurs prior to chromatin sonication and immunoprecipitation. The combination of these two alterations demonstrably improves the efficiency of the method and the signal to noise ratio.
Hi-C: The Next-Gen Chromosome Conformation Capture Method
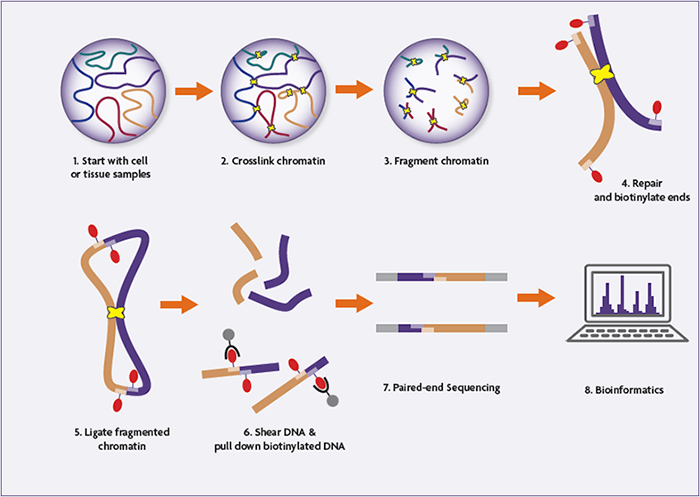
Because Hi-C assays enable the investigation of genome-wide chromatin interactions, it is central to the ongoing efforts to understand the 3-dimensional organization of the genome and the physiological role it plays.
In the decade since its debut, the Hi-C methodology has become the most popular assay for high-throughput analysis of higher-order chromatin structure and has been crucial for revealing the complex 3-dimensional chromosome landscape of loops and topologically associated domains (TADs). Hi-C has also been useful for understanding the relationship between chromatin organization and gene expression and observing changes in disease state architecture.
Want to keep learning about Hi-C? Check out the interview with Erez Lieberman Aiden, one of the scientists that originally developed Hi-C, on our Epigenetics Podcast
What is the Hi-C Protocol & How Does it Work?
Hi-C is an attractive option for querying genome-wide chromatin interactions as well as the differences found in response to signaling, differentiation, or disease. Below, we outline the principles and details of the general Hi-C protocol.
- Cells are fixed with formaldehyde (or another fixative), crosslinking DNA-DNA and DNA-protein interactions.
- The cells are lysed, and the crosslinked DNA is digested with a restriction endonuclease.
- The resulting 5’ overhangs are filled in, incorporating a biotinylated nucleic acid.
- Blunt-end ligation is performed under very dilute conditions, favoring the ligation of crosslinked strands of DNA. This generates a circular, hybrid DNA strand which is biotinylated at the ligation junction.
- The crosslinking is reversed, and the samples are sonicated/sheared to between 300 and 500 base pairs.
- Streptavidin beads are used to pull down the biotinylated linear fragments, enriching the sample for genuine interactions.
- Paired-end adaptors are ligated to the fragments, and the library is amplified and sequenced using high-throughput next-generation sequencing platforms.
- The resulting sequences represent interacting fragments, which are mapped back onto the genome for identification.
Learn More About Active Motif's End-to-End Hi-C Service
Pros & Cons of Hi-C
At present, Hi-C is the most extensively utilized chromosome capture method thanks to a long list of advantages and applications. As discussed above, Hi-C is the standard for assessing changes in genome-wide interactions, since it observes all interactions amongst all loci in an unbiased fashion. Hi-C also provides an unbiased approach. The Hi-C “all-vs-all” approach makes it particularly useful for applications involving genome-wide changes to chromosome topology.
The limitations of Hi-C center predominantly around issues of resolution of the assay. In general, Hi-C is not well suited for probing short distance interactions such as intra-TAD interactions. This issue can be resolved by using a restriction enzyme with a higher digestion frequency (i.e. a 4 base pair cutter instead of 6 base pairs) but working at this high resolution over the large breadth of the genome can be cost-prohibitive.
Hi-C vs. 3C, 4C, and ChIA-PET
Many of the issues that plague the chromosome conformation capture methods can be avoided by thoughtful and careful choice of method. Choosing a method in line with the goals of a study goes a long way towards generating clean and useful datasets. Here, we outline a basic comparison of the more commonly used chromosome capture methods.
Hi-C vs. 3C
3C and Hi-C are at opposite ends of the chromosome conformation capture spectrum. While 3C can only investigate interactions between two known loci, Hi-C captures genome-wide interactions across all loci. Because 3C primers are designed against a known locus, it requires prior knowledge of the genes being investigated and therefore introduces a bias in what interactions will be assayed. On the contrary, Hi-C is unbiased and high-throughput. 3C may be more suitable for small-scale interactions such as looping and intra-TAD interactions, but Hi-C is the clear choice for the identification of genome-wide novel interactions or changes to genome-wide topology.
Hi-C vs. 4C
Both the 4C and Hi-C methods can be used to identify novel chromatin interactions and changes to genome topology, but 4C takes the viewpoint of a single locus as opposed to the genome-wide powers of Hi-C. The more local viewpoint of 4C, like 3C, comes with an inherent bias in choosing what region to observe. While the limited throughput of 4C may sound like a disadvantage, it provides a more cost-effective way to observe a known or clinically relevant locus in greater depth.
Hi-C vs. ChIA-PET
Both Hi-C and ChIA-PET are genome-wide methods, but ChIA-PET generates a more protein-centered view of the chromosome. Both methods have the advantage of a biotin pull-down step which enriches the relevant sample population, improving the signal to noise ratio. ChIA-PET also suffers from bias issues, since the method isolates DNA bound to a specific protein, and the researcher must choose which protein to immunoprecipitate. Hi-C, on the other hand, measures all DNA-DNA interactions across the genome without requiring a choice of viewpoint, region, or protein.
Discoveries Enabled by Hi-C
If the long list of Hi-C advantages isn’t proof enough of its usefulness, Hi-C also comes with a lengthy and impressive resume. Aside from its integral role in the early discoveries of various 3-dimensional chromosome architecture elements, Hi-C has also been the method of choice for more recent noteworthy discoveries.
As previously discussed, one of the strengths of Hi-C is the ability to look at global changes to genome architecture. A 2019 study published in the journal Nature utilized Hi-C for studying changes to mouse chromatin structure during the transition from mitosis to G1. Hi-C data showed that A/B compartments are rapidly formed after mitosis is complete, and delineated both the expansion of these compartments as well as the accumulative formation of TADs over time.
An in situ Hi-C system was used to show chromatin reorganization post-fertilization in mouse oocytes. TADs, loops, and compartments were observed to be rearranged during this transition from oocyte to zygote.
In the same vein and using a similar system, Hi-C was employed to compare the genomic architecture of embryonic versus neuronal Drosophila cells, and then for identifying conserved and divergent TADs. This study also observed changes to DNA occupancy and architectural protein complexes at TAD borders, tracking with changes in gene expression.
What’s Next for Chromosome Conformation Capture Assays?

Since the debut of the 3C chromosome conformation capture assay in 2002, the available methods have undergone several iterations of refining and these assay updates have improved their resolution, throughput, specificity, and efficiency.
Like many other areas of research, the chromatin architecture field is moving towards single-cell studies, which may uncover regulatory features currently masked by pooled population studies. Further development of these methods will provide a higher resolution view of the 3-dimensional genome, which can be applied to studying the dynamic genomic landscape in unique cell types and in response to biological events.
Additionally, improved tools for visualizing the 3-dimensional genome and integration with other -omics platforms may provide better insight into the relationship between genome topology and gene expression.
Lastly, it is hopeful to presume that a better understanding of genome topology will reveal how this organization is perturbed in human disease and offer up fresh perspectives for treatment.
Summary: 3-D Organization of the Genome is the Next Frontier in Epigenetics
As we gain an appreciation for the complex and dynamic entity that is the 3-dimensional genome, methods like Hi-C are allowing researchers to address novel research questions and contributing to unveiling a more complete view of the genome. Large-scale efforts are being directed towards mapping and annotating the 3-dimensional genome and understanding the role of genome topology in gene regulation. This field of research holds much promise for genomics-related research, and these new approaches are expanding the types of questions researchers can ask, ultimately aiming us towards a more detailed and holistic view of the genome.
Want to keep learning about Hi-C? Check out the interview with Erez Lieberman Aiden, one of the scientists that originally developed Hi-C, on our Epigenetics Podcast
<< Back to MOTIFvations Blog Home Page




