<< Back to Resources Home Page
完全ガイド:エピジェネティクス入門

目次
エピジェネティクスとは、環境や行動、その他の要因がどのようにゲノムに影響を与えるかを研究する学問です。これらの影響は、遺伝子そのもののDNA配列を変えることなく、遺伝子の働きやその他の生物学的プロセスを調節します。エピジェネティクスのメカニズムは、DNAやタンパク質(特にヒストン)、RNAの化学的修飾によって起こり、これらの修飾は多くの場合、可逆的であり、遺伝することも知られています。
この記事では、エピジェネティクスの基本概念や歴史について紹介し、エピジェネティックなプロセスで重要な役割を果たす因子や要因について、それらがどのように生物学的プロセスに影響を与えるか解説します。
エピジェネティクスとは何か?
エピジェネティクスの定義は、1942年にConrad Hal Waddingtonによって初めて提唱されて以来、議論の的になっていますが、この言葉自体は、「外側」や「上に」という意味を持つギリシャ語の接頭辞「epi-」と、遺伝学として知られる「genetics」の組み合わせに由来しています。この組み合わせた語句は、遺伝子の継承に加えて、もう1つ別の意味として、受け継がれる第一の情報(DNA配列)の外側に第二の情報(修飾)があることも示唆しています。現在、多くの人が、エピジェネティクス(後成遺伝学)とは、DNAの一次配列に変化が生じることなく、細胞や生物の世代を超えて受け継がれる遺伝子発現の違いを研究する学問であると認識しています。
エピジェネティクスの研究は新しい分野とされていますが、その実例は何千年も前から私たちの身の回りに存在しています。例えば、今から数千年前、ラバ(mule)※の繁殖家たちが、オスのロバ(donkey)とメスの馬(horse)を交配させるとラバが生まれ、オスの馬とメスのロバを交配させるとヒニー(hinny)が生まれることを発見したとき、実はエピジェネティクスと遭遇していたのです。ヒニーはラバよりもおとなしく、やや臆病で神経質と言われますが、体格は馬(父)よりもロバ(母)に近く小さいものの脚力が強いことが知られています。ラバもヒニーも馬とロバの交配から生まれますが、どちらの親から遺伝子を受け継ぐかにより、その特徴は異なります(※訳注:日本語では「ラバ」という場合、通常はミュール(父親がロバ、母親が馬)を指すことが多いようです。一方で、父親が馬で母親がロバの場合は、日本語にはあまり特別な呼称がなく、「交配種」や「ヒニー」とそのまま呼ばれることが一般的なようです)。
他にもエピジェネティクスの代表的な例として、蝶が蛹の中でアオムシから成虫へと変化する様子があります。アオムシは十分に餌を蓄えると、足場となるように糸を張りながら自身を固定し、柔らかいアオムシから成虫に形態を変化させて、その後、複眼、翅、脚を持つ蝶になります。この2つの段階における形態はまったく異なるにもかかわらず、アオムシと成虫(蝶)の遺伝子は両者で変わることはありません。唯一の違いは、遺伝子がどのように発現したか、それだけの違いです。
私たちの体は、最初はたった1つの細胞、つまり精子と卵子が結びついてできる受精卵 (接合体)から始まります。この受精卵が胚となり、やがて数十兆個もの細胞を持つ人間の体へと成長していきます。細胞分化として知られるこの過程において、受精卵は多能性幹細胞へと発達し、体内のあらゆる器官を生み出すことができるようになります。人体を構成するすべての細胞は、すべて1つの受精卵を祖先とするため、同一のDNA配列を持ちます(生殖細胞や免疫細胞を除く) 。体のさまざまな組織や構造を形成するための情報をすべて含んだ同じDNA配列を持つにもかかわらず、個々のの細胞は異なる分化過程をたどり、神経や筋肉、さまざまな内蔵など多様な器官を形成していきます。
こうしたプロセスは、DNA配列だけでは説明がつかないため、第二の制御が必要であると考えられました。そこで登場するのがエピジェネティクスであり、特定の遺伝子発現がDNA配列以外の何によって調節されるのか、そのメカニズムを理解する学問です。
エピジェネティクスの歴史
1942年に「エピジェノタイプ」という言葉が初めて提唱したのがConrad Hal Waddingtonです。彼は、「遺伝子型と表現型の間には、非常に複雑な発生過程が存在する」と述べています。この考え方は、1650年頃にWilliam Harveyが提唱した「エピジェネシス(epigenesis)」という古典的な概念に基づいています。
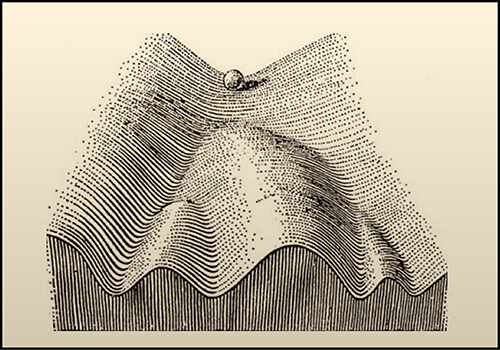
その後、WaddingtonはD. melanogasterの胚を用いたいくつかの実験を行った後、 1957年に「エピジェネティック・ランドスケープ 」というモデルを提唱しました。右のイラストは、山や谷のある斜面をボールが転がって進む様子を、細胞が分化過程においてどのように運命の分かれ道を進み、その系譜が決定されるかにたとえています。このモデルでは、分化の方向の基礎となる遺伝子の働きが「風景(ランドスケープ) 」を形づくり、それに依存して細胞の運命 (ボールの進行方向)はいくつかの可能性をもちながらも一定の範囲内に制限され、細胞系譜が決まることを示しています。
その1年後、David Ledbetter Nanneyによって2つの細胞制御システムが提唱されました。1つ目は、DNAの一次配列にコードされた遺伝子に対応する「特異性のライブラリー(library of specificities)」であり、もう1つは、「補助機構(auxiliary mechanism)」と呼ばれるシステムで、特定の細胞にどのような特異性を発現するかを決定するものでした。彼は、話をよりシンプルにするため、「遺伝システム(genetic system)」と「エピジェネティック・システム(epigenetic system)」という言葉を使うことを提案し、Waddingtonの論文をふまえて、エピジェネティックなプロセスが遺伝システムに依存していることを強調しました。
クロマチンの発見
「エピジェネティクス」の分子機構が注目されるようになったのは2000年代に入ってからですが、それを理解するための基礎はかなり以前から築かれていました。19世紀後半、Walther Flemmingは細胞分裂前と分裂中の細胞核の構造を染色して研究しています。これが画期的な研究へとつながりますが、彼はそこでヒト染色体のおそらく最初の画像を発表しました。
「クロマチン(chromatin)」という言葉を最初に作ったのは彼であり、染色されやすい核内の糸を表現しました。一方で、染色できない構造のことを 「アクロマチン(achromatin)」 と呼びました。彼はこれらの構造が何かについて正確には理解していませんでしたが、何か重要なものであるということには気づいていたようです。彼は 『...化学的性質が明らかになるまで、クロマチンという言葉は使用できるだろう。そしてその間、細胞核内で容易に染色される物質を指す言葉として用いられるだろう』と記しており、この「クロマチン」という言葉は、今日でも、細胞内に見られるDNAとタンパク質の糸のような複合体を表す言葉として使われています。
その後まもなく、Albrecht Kosselにより、核内DNAの折りたたみ構造の調節に関連するアルカリ性タンパク質の一群として、ヒストンが発見されました。彼のよく知られた功績としては、核酸を構成する有機塩基の発見が挙げられますが、彼はガチョウの赤血球の核から酸抽出可能なペプトン様物質を単離したことも報告しています。当時は、その役割は明確ではありませんでしたが、彼はそれが核酸と結合している可能性を示唆し、「ヒストン」と命名しました。
1960年代には、ヒストンがエピジェネティクスへの関与を示唆する最初の報告がなされました(Allfrey, Faulkner, and Mirsky, 1964, PNAS)。この論文には、ヒストンのアセチル化とRNA合成の制御との関連について記述されています。また、RNA合成はアセチル化されたヒストンよりも、ネイティブ状態の方が抑制されることを示唆するデータが示されました。彼らは、ヒストンのアセチル化が「RNA合成のオン・オフを切り替える手段」である可能性を示唆しました。ヒストンが遺伝子発現を制御するかもしれないという仮説は、エピジェネティックなメカニズムをその後理解する上で非常に重要な一歩となりました。
それからまもなく、ヒストンのコアにDNAが巻き付いた染色体の基本構造単位であるヌクレオソームが初めて可視化されました。1973年、AdaとDon Olinsは電子顕微鏡を駆使してクロマチンの中に直径約70 Å (オングストローム)ほどの球状粒子を撮影することに成功し、これを“ν bodies”と命名しました(訳注:英語のvではなく、ギリシャ文字のニュー)。さらに、これらの球状粒子が幅約15 Åの鎖で結ばれ、「紐付きビーズ」のように見えることも明らかにしました。1975年には、Roger D Kornbergがこのクロマチンの基本構造をさらに詳しく解析し、『…4種類の主要ヒストン2個ずつと約200塩基対のDNAを単位とする繰り返し構造である。クロマチン線維は、このような単位が多数集まり、柔軟に結合した鎖を形成している』と述べました。この構造単位はその年、現在の「ヌクレオソーム」と呼ばれるようになりました。
ヌクレオソームの発見と定義は、クロマチンに対する認識を一変させました。現在では、クロマチンは、球状のヒストンコアの外側にDNAが巻きついた構造を持ち、外側から核内タンパク質との相互作用や結合が可能な状態にあることが知られています。
その後ほどなくして、ヌクレオソームの結晶構造を解明する競争が始まりました。最初のヌクレオソーム構造は、1984年にTimothy Richmondによって7.0 Åの分解能で決定されました。そして1997年、ついにKarolin Lugerらがヌクレオソームの結晶構造を2.8 Åの分解能で決定し、各ヒストン8量体がどのように組み合わさり、その周囲に146塩基対のDNAが『スーパーヘリックス』状に組織化されているかを明確に示しました。そして複数のヒストン-ヒストン相互作用とヒストン-DNA相互作用が決定され、ヒストンテールに存在する特定のアミノ酸がDNAにどのように相互作用し、影響を与えるかを理解する道が開かれたのです。
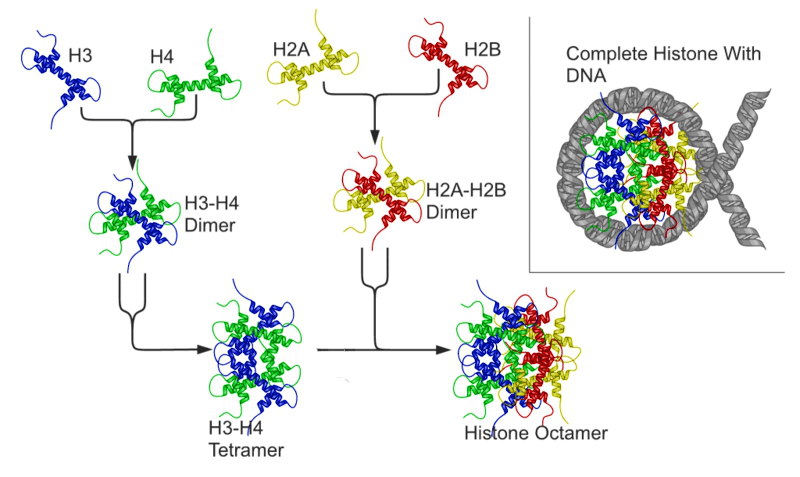
Image credit: Nucleosome_structure-2.png - CC BY-SA 4.0
クロマチンに関する重要な多くの知見は、2001年にThomas JenuweinとC. David Allisが発表した “Translating the Histone Code”という画期的な総説論文に見事にまとめられています。そこでは、ヒストンテールの翻訳後修飾が、クロマチン関連タンパク質と相乗的あるいは拮抗的な相互作用につながる可能性を示しています。 さらには、これらの異なるヒストンマークの相互作用や組み合わせが、転写的に活性化(ユークロマチン)または不活性化(ヘテロクロマチン)したクロマチン状態を決定づけ、異なる局所環境に導く可能性があることについても提唱しました。そしてそこでは、クロマチンのリモデリングはエピジェネティックな制御機構として極めて重要であり、『細胞の運命決定や正常な発生、および病的な発生に多大な影響を及ぼす』と提唱されています。
DNAメチル化の解明
歴史的に、DNAメチル化の研究はヒストンの研究と並行して進められていましたが、真核生物におけるDNAメチル化は、1948年にRollin Hotchkissによって初めて発見されました。彼はペーパークロマトグラフィーを用いて、子牛胸腺由来DNAのシトシンの一部が修飾されていることを発見し、それが5-メチルシトシン(5-mC)であるという仮説を立てました。というのも、チミン(別名:メチルウラシル)はウラシルがメチル化されているのと同様だと考えたからです。
しかし、実際にDNAメチル化とエピジェネティクスの関係が明らかになるまでには1970年代半ばまでかかりました。 1975年、AD RiggsはX染色体の不活性化と配列特異的DNAメチラーゼを結びつけるモデルを提唱しました。X染色体の不活性化は、メスがX染色体を2コピー受け取るのに対し、オスは1コピーしか受け取れないために起こります。メスの細胞が多くのX連鎖遺伝子産物をオスの2倍ずつ産生するのを防ぐには、X染色体の一方を不活性化しなければなりません。どちらを不活性化するかは細胞ごとにランダムに決まりますが、細胞の寿命が尽きるまでは不活性化されたままです。不活性化されたX染色体は、一部を除き高レベルのDNAメチル化により、遺伝子の発現を抑えられています。
同じ年、R. HollidayとJ.E. Pughは、バクテリアで知られていたDNA修飾の特徴に基づき、真核生物におけるDNA修飾酵素が、発達過程の遺伝子活性を制御するであろうメカニズムについて提唱しました。当時、真核生物に特異的なDNA修飾酵素が存在するという直接的な証拠はまだありませんでしたが、彼らは、なぜDNA修飾酵素を実験的に探索する意義を次の例を挙げて主張しました。 第一に、DNAのメチル化がランダムではないこと、第二に、メチル化されたCpG配列が頻繁に観察される一方で、DNA中における偶然の出現頻度としては低確率であること、第三に、 ウニの胚においてDNAメチル化酵素が確認されていたことです。これらのことから、DNAのメチル化が細胞内で能動的に制御されている可能性が高いと考えたのです。
1980年代から1990年代にかけて、Adrian BirdがCpGアイランドにおけるDNAメチル化の特徴について先駆的な研究を行いました。彼による脊椎動物のメチル化遺伝子領域の同定は、メチル化感受性制限酵素によるゲノムDNAの切断パターンを利用することにより可能となりました(※訳注:DNAメチル化によりDNA切断活性が阻害される酵素。具体的にはHpa II)。彼は、メチル化された領域が通常、1-2 kbpの長さからなる「島(island)」としてゲノム上に存在し、約100 kbpに1ヶ所の割合で離散的に分布していることを発見しました。これらは現在 、「CpGアイランド」 として知られています。彼はさらに、CpGアイランドは通常、遺伝子のプロモーター領域またはその近傍で完全にメチル化されているか、あるいは全くメチル化されていないという、どちらかの状態で見つかることを発見し、遺伝子制御と転写に関与していることを示唆しました。
その後数年間は、DNAメチル化の内因性パターンを決定すること、そして、これらのパターンが細胞や生物個体における世代間でどのように受け継がれていくかに焦点が当てられてきました。その結果、DNAメチル化には2種類あり(de novoメチル化(新たなメチル化)、および維持メチル化(既存のメチル化領域の維持))、多くの異なる酵素がそれぞれ関与していることが判明しています(後述)。
エピジェネティック修飾の種類
核内における遺伝物質の構成は、DNAへのアクセスを必要とする全てのプロセスに重大な影響を及ぼします。クロマチンは動的な構造体であり、クロマチンを構成するタンパク質やDNAに共有結合的な修飾が加わると、クロマチンの構造が変化します。中でも最も研究されているのは、DNAのシトシンにおける共有結合修飾と、ヒストンテールのリジン、アルギニン、セリン、スレオニンにおける翻訳後修飾(PTM: post-translational modification)です。
このような修飾は、タンパク質とDNAの相互作用を変化させ、クロマチンの物理的状態を変化させます。いくつかの修飾は、ユークロマチンとして知られる、ゆるく開いたクロマチン構造を作り出し、転写因子が遺伝子上流のプロモーターへ結合することを可能にします。これにより、RNAポリメラーゼII (RNA pol II)がDNAに結合できるようになります。ユークロマチンはゲノム中の最も転写活性の高い領域を形成していますが、これとは対照的に、ヘテロクロマチンとして知られる閉じたクロマチン状態では、転写因子によるDNAへのアクセスが制限され、ゲノムの不活性領域を形成します。
DNAとヒストンの共有結合修飾は、さまざまなタンパク質により厳密に制御されています。修飾を加えるタンパク質もあれば、修飾を取り除くタンパク質もあります。このようにして、遺伝子発現は環境の変化や有糸分裂のいずれに対しても、必要に応じて変化して適応します。このような変化は塩基配列を変えることなく有糸分裂後の娘細胞に受け継がれ、あるいは生殖細胞系列を通じて次世代に選択的に受け継がれていきます。
ヒストンH3K4メチル化 -プロモーターマーク
ヒストンH3K4はトリメチル化される部位として最もよく知られており、活性化したプロモーターの目印となるため、これをプロモーターマークと呼ばれます。しかし、H3K4はアセチル化修飾されることもあります(後述)。
H3K4me3 (ヒストンH3の4番目のリジンのトリメチル化)は、活性化したプロモーターの転写開始点(Transcription Start Site; TSS)に局在し、WDR5ヒストンメチルトランスフェラーゼ酵素によりメチル化されます。一方、H3K4の脱メチル化酵素にはKDM5B/JARID1Bなどがあり、H3K4の各種メチル化状態を脱メチル化して元に戻すことができます。
H3K4me1(ヒストンH3の4番目のリジン上のモノメチル化)は、待機状態のエンハンサーと活性型エンハンサーに存在するヒストンマークとして知られています。H3K27acと一緒に観察されることが多く、両方が存在する場合は活性型エンハンサーであり、H3K4me1のみが存在する場合は待機状態にあることが知られています。ライター(writer)として働くメチル化酵素はKMT2CとKMT2B(それぞれMLL3とMLL4とも呼ばれる)です。リジン脱メチル化酵素であるKDM1A/LSD1は、このマークのイレーサー(eraser)として働きます。
H3K4me3の濃縮領域(転写開始点: TSS)のすぐ上流、すなわち活性化遺伝子のプロモーター上には、H3K4acが見られます。
ヒストンH3K9メチル化 - ヘテロクロマチンマーク
ヒストンH3K9の修飾は、閉じたクロマチンの維持に関わるH3K9me2およびH3K9me3が最もよく知られており、ヘテロクロマチンマークと呼ばれています。しかし、H3K9はアセチル化されることもあり、その場合、活性型の遺伝子プロモーターの目印となります。
H3K9me2とH3K9me3(ヒストンH3の9番目のリジンのジメチル化またはトリメチル化)はヘテロクロマチンに見られる特徴です。これらのヒストンマークは、分化過程において細胞系譜の方向性を維持する上で極めて重要であり、ゲノムの特定の部分が転写されるのを制限し、細胞の再プログラミングに対する障壁として働きます。
ヒストンH3K9は、SETドメイン含有メチルトランスフェラーゼSETDB1と、SUV39H1およびSUV39H2によってメチル化されます。それらのメチル化されたヒストンマークの主なリーダー(reader)はHP1 (heterochromatin protein 1)です。HP1はH3K9me2やH3K9me3に結合すると、より抑制的なヒストン修飾因子をリクルートします。しかし、多能性を求められる幹細胞では、幹細胞へのリプログラミング・複製に関わる遺伝子座におけるH3K9me2とH3K9me3は、それぞれヒストン脱メチル化酵素KDM3AとKDM4Cにより消去されます。
H3K9acは活性型の遺伝子プロモーターに見られますが、H3K9me1は活性化遺伝子の転写開始点(TSS)付近に見られます。
ヒストンH3K27のメチル化とアセチル化 – 2面性を持つヒストンテール残基
H3K27 (ヒストンH3の27番目のリジン)の翻訳後修飾は、今日、制御エレメントがオンかオフかを示す最も信頼性の高いヒストンマークとして知られており、アセチル化されるとオープンクロマチンを示し、トリメチル化されるとクローズドクロマチンを示します。
H3K27me3は、抑制型エンハンサーやプロモーターに局在し、ヘテロクロマチンを示します。このマークの最も一般的なライターはメチル化酵素のEZH2であり、イレーサー(eraser)には脱メチル化酵素であるJMJD3とUTXがあります。H3K27me3のリーダーは、ポリコーム複合体(Polycomb Repressive Complex)のPRC1とPRC2です。PRC2が遺伝子サイレンシングを開始し、PRC1は、分化後に必要とされるクローズドクロマチンの維持に寄与します。このマークのイレーサーはリジン脱メチル化酵素であるKDM6Aです。
H3K27ac(ヒストンH3の27番目のリジン上のアセチル化)は豊富に存在しますが、おそらく最も広範に観察される活性化領域およびオープンクロマチンのマークとして知られています。アセチル化されたH3K27は、活性化型プロモーターや活性型エンハンサーに存在し、アセチル化酵素としてはp300やCBPが、脱アセチル化酵素としてはHDAC1やHDAC2が知られています。
ヒストンH3K36のメチル化 - 活発な転写とDNA複製・組換え・修復マーク
H3K36のメチル化は、活発な転写、DNA損傷の修復、複製、組換えの部位で見られます。 SETドメインタンパク質群が、この残基のモノメチル化、ジメチル化、あるいはトリメチル化のライターとして働きます。H3K36メチル化のリーダーとしては、PHF (Plant Homeodomain Finger)タンパク質群、Chromoドメインタンパク質、Tudorドメインタンパク質群、PWWP (Pro-Trp-Trp-Pro)ドメインタンパク質群が知られています(訳注: PHFタンパク質群に含まれるPHF1やPHF19は、どちらもTudorドメインも有していますが、PHFファミリーに属さないTudorドメインタンパク質TDRD3もリーダーの1つとして知られています)。KDM (ヒストンリジン脱メチル化酵素)ファミリータンパク質は、このヒストンマークのイレーサーとして機能します。興味深いことに、H3G34 (ヒストンH3の34番目のグリシン)は、いくつかの小児がんにおいて変異しており、この変異がH3K36のメチル化を阻害することが知られています。これにより、DNA損傷修復やH3K36のメチル化が介する他の機能が阻害されます。
H3K36me2 (ヒストンH3の36番目のリジンのジメチル化)は、転写開始に関与しています(転写開始点でSETD2により付加)。 SETD2はリン酸化されたRNA Pol IIと相互作用すると、ヒストンH3をH3K36me3までメチル化し、RNA Pol IIの伸長反応が始まります。H3K36のジメチル化は遺伝子間領域(intergenic regions)においても役割を果たしています。 H3K36me2は別のSETドメインタンパク質ヒストンメチルトランスフェラーゼNSD1により書き込まれ、このマークはDNAメチルトランスフェラーゼDNMT3Aをリクルートし、この領域におけるDNAメチル化の維持に寄与します。NSD1のハプロ不全は、ソトス症候群(Sotos syndrome)の原因として知られており、遺伝子間領域におけるH3K36のジメチル化の減少と、それに伴うDNAメチル化の欠損を引き起こします(訳注:ハプロ不全とは両親由来の2コピーの遺伝子のうち、1コピー分しかタンパク質が発現せず、機能的に不十分な状態)。
H3K36me3 (ヒストンH3の36番目のリジン上のトリメチル化)は、前述のように、RNA Pol IIと結合した後にライターであるSETD2により付加されます。このマークは、転写伸長の役割を果たします。 なお、H3K36のメチル化は、ヒストン脱メチル化酵素であるKDM2A-BとKDM4A-Dにより消去されます。
H3K36me1は、まだ詳細には解明されていませんが、H3K36のジメチル化およびトリメチル化修飾と同様に、遺伝子の活発な転写に関連することが知られています。このマークのリーダーとしてはヒストン脱アセチル化酵素複合体RPD3である可能性が示唆されています。
H3K36acは植物において、活発に転写されている遺伝子の5‘末端に見られる修飾として特徴づけられています。
DNAメチル化とメチル化バリアント
DNAメチル化は、遺伝子発現とゲノム構成の重要な調節因子です。真核生物のDNAのメチル化はシトシンで起こり、DNAメチル化酵素(DNMT: DNA methyltransferase)により5-メチルシトシン(5-mC)に変換されます。DNAのメチル化は、ほとんどがCpGジヌクレオチドにのみ現れます。これらの連続したヌクレオチドは、哺乳類のゲノムでは比較的にまれであり、CpGアイランドと呼ばれる場所に集まる傾向があります(訳注:後述するシトシンの脱アミノ化に起因するCpG出現頻度の低下(CG抑制)により、CpGアイランド以外における出現頻度は約1%しか存在しません) 。遺伝子プロモーターの約60%はCpGアイランドと関連しており、遺伝子が発現するときはメチル化されていません。遺伝子プロモーターのCpGメチル化は通常、転写抑制と密接な関係があります。
DNAメチル化は、de novo DNAメチル化とDNA維持メチル化という2つの異なる酵素プロセスで行われます。
de novo DNAメチル化は、それまでメチル化されていなかったDNAにメチル基を付加するもので、主に胚性幹細胞(ES細胞)、初期発生胚、発生中の男女生殖細胞に存在しますが、成人の分化した体細胞ではほとんど抑制されています。de novo DNAメチル化は、DNMT3A、DNMT3Bにより生じ、配偶子形成期および子孫における父方とと母方の対立遺伝子の発現に相違を生じるゲノムインプリンティングの確立に重要な役割を果たしていると考えられています。
他方、DNA維持メチル化は、細胞のDNA複製サイクルごとにDNAメチル化を維持するために重要と考えられています。これは主に、DNMT1により実行されます。成体の細胞分裂におけるDNA複製では、2本鎖DNAがほどけて1本鎖DNAが生じます(親鎖)。これを鋳型とする新生鎖はメチル化されていないため、親鎖由来の1本鎖のみがメチル化されたヘミメチル化DNAが生じます。DNMT1は、これらのヘミメチル化したCpGに結合して新生鎖のシトシンをメチル化します。これにより、有糸分裂を通じて確立されたCpGメチル化パターンが維持されます。この仕組みがなければ、DNAのメチル化は細胞分裂を繰り返すうちに希薄になり、最終的には失われてしまうことになってしまいます。
メチル化されたDNAは元に戻すこともできます。メチルシトシンのジオキシゲナーゼファミリーに属するTET (ten-eleven translocation)酵素群は、DNA脱メチル化において中心的役割を担っています。これらの酵素は5-mCを酸化して5-hmCに変換し、さらに5-ホルミルシトシン(5-fC)、そして5-カルボキシシトシン(5-caC)へと段階的に酸化を触媒します。その後、5-fCと5-caCは、チミンDNAグリコシラーゼ(TDG: thymine-DNA glycosylase)による除去から始まる、塩基除去修復(base excision repair)機構により非メチル化シトシンに置換されて元に戻ります。
ある種の酵素がDNAメチル化コードの書き込みと書き換えに関与する一方で、このエピジェネティック・マークの読み取りには、他のタンパク質が関与しています。Methyl-CpG-binding domain タンパク質(MBD)は、対称的にメチル化された1つ以上のCpGペアを含むDNAに特異的に結合します。各MBDは、メチル化CpGペア周囲の約12ヌクレオチドをカバーすることができる構造タンパク質として働き、さまざまなヒストン脱アセチル化酵素(HDAC)複合体やクロマチンリモデリング因子をリクルートし、クロマチンの凝縮とそれに続く転写抑制を誘導します。
DNAのメチル化は、胚発生、ゲノムインプリンティング、X染色体の不活性化、遺伝子発現の制御など、多くの細胞機能に重要です。正常なDNAメチル化パターンの変化は、がん、先天性疾患、心血管系疾患など、さまざまな疾患過程と相関することが示されています。がんでは、プロモーター領域におけるCpGアイランドの過剰なメチル化により、細胞分裂を抑制する重要な遺伝子の発現が失われることがあります。また、シトシンは脱アミノ化されるとチミンへと変わりますが、メチル化シトシンは、非メチル化シトシンよりも自発的脱アミノ化を起こすため、ゲノム上で変異しやすくなります。その結果、本来G-C対となる部位にミスマッチのG-T対が生じるため、これらは修復されなければなりません。もし、ゲノム複製前に修復されなければ、CからTへの突然変異は永久にゲノムに書き込まれることになってしまいます。
DNMTやTET、MBDなど、DNAメチル化状態を変化させたり、読み取ったりするタンパク質の変異や発現変化も、多くの疾患につながります。例えばMethyl-CpG Binding Protein 2 (MECP2)タンパク質の変異はRett症候群を引き起こし、DNMT3AとTET2の両タンパク質の変異はがんの発生と関連しています。
DNAのメチル化は、老化プロセスにも強く関係していることが示されています。そして、加齢にともなうDNAメチル化パターンは、組織や細胞において予測することが可能であることがわかってきました。Steve HorvathとKenneth Rajが、2018年にNature誌に発表した論文では、DNAメチル化データを組織の生物学的老化にうまく適応させることにより、一種の『エピジェネティック・クロック』を作り出すことができたと述べられています。今後、正常なDNAメチル化パターンに対する理解が深まり、それを細胞内で測定するための新しいツールが登場すれば、DNAメチル化に関連する疾患の診断や治療につながる可能性があります。
ヒストン修飾
ヌクレオソームはクロマチンの基本単位であり、8個のヒストンタンパク質とそれらに巻きついた147 bpのDNA部分から構成されています。このヒストン8量体のコアは、ヒストンタンパク質H2A、H2B、H3、H4によって形成されています。 これら約18 kDaのヒストンタンパク質群は、進化的によく保存されており、それらは“helix turn helix”モチーフを特徴とした構造をとっています。ヌクレオソームを形成するため、H3とH4はヘテロ二量体を形成し、それがさらに二量体化してH3-H4四量体となります。これが最終的に2つのH2A-H2Bヘテロ二量体と会合することにより8量体を形成します。
ヒストンはプラスに帯電したアミノ酸残基を多く含むため、DNA骨格に含まれるマイナスに帯電したリン酸基と安定な静電相互作用を形成するします。このようにしてDNAがヒストン8量体の周りをしっかりと包み込むことができます。これらのプラスに帯電した残基は、各ヒストンのN末テールに多く存在しています。このテール部は8量体コアから突出しているため、ヌクレオソームの外側からアクセスできます。これにより、これらのテールはヌクレオソーム間、およびヌクレオソーム内の重要な相互作用において役割を果たしています。
ヒストンテールの修飾は主にリジン残基(K)で起こりますが、アルギニン(R)、スレオニン(T)、セリン残基(S)でも起こります。リジンとアルギニン残基はメチル化またはアセチル化され、セリン残基とスレオニン残基はリン酸化されます。
リジン残基がアセチル化されると、正電荷が失われます。これによりDNAとの結合が弱まり、クロマチン構造がよりオープンな状態になります。ヒストンのアセチル化は多くの細胞内プロセスに関与しますが、最も一般的な機能は転写の活性化です。ヒストンのアセチル化は遺伝子のプロモーター領域に多く見られます。これらの領域のクロマチン構造をゆるめることにより、転写因子がアクセスできるようになり、結果的に遺伝子発現が増加します。
一方で、リジン残基のメチル化の機能を予測するのはやや困難です。ヒストンテールのどのリジン残基がメチル化されるか、あるいはそれがモノメチル化、ジメチル化、トリメチル化されるかにも影響を受けます。その結果、転写が抑制されることもあれば、活性化されることもあります。ヒストンテールとDNA間の化学的結合を弱めるようなメチル化反応は、アセチル化と同様に転写を促進します。なぜなら、これによりDNAがヌクレオソームからほどけ、転写因子タンパク質やRNAポリメラーゼがDNAにアクセスできるようになるからです。リジン残基とアルギニン残基のメチル化は、クロマチンにおける転写活性領域と不活性領域形成の主な決定因子として機能し、発生過程におけるゲノムの適切なプログラミングに非常に重要とされています。
セリンおよびスレオニン残基のリン酸化の詳細はわかっていませんが、DNA修復と有糸分裂において特に重要であることが示されています。例えば、ヒストンH3のテールにある10番目のセリン (Ser10)と28番目のセリン (Ser28) のリン酸化は、染色体凝縮が誘導される有糸分裂初期に生じます。
4つのコアヒストン(H2A、H2B、H3およびH4)すべてのヒストンテールにおける修飾は、遺伝子制御に関与しています。その中でもヒストンH3とヒストンH4のテールは特に重要であることが示されています。ヒストンH3のN末テールには修飾されることが既知のアミノ酸として、2番目にアルギニン、4、9、14、18、23、27、36、56、79番目に9個のリジン残基、28番目にセリン残基が存在します。そしてヒストンH4には3番目にアルギニン、5、8、12、16、20番目に5つのリジン残基、1番目にセリン残基があります。
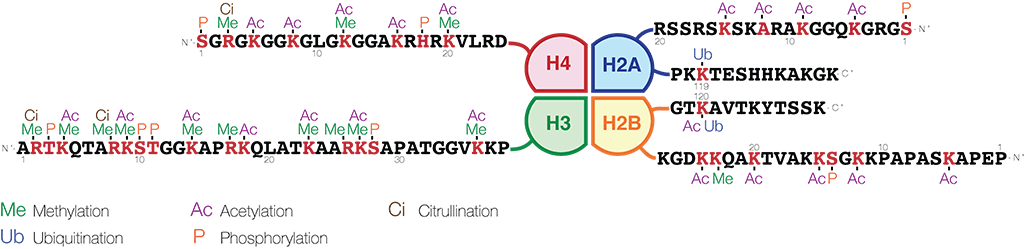
Image credit: https://en.wikipedia.org/wiki/Histone - CC BY-SA 4.0
ヒストン修飾のリーダー、ライター、イレーサー
T様々な組み合わせで修飾されたヒストンの可能性は無限にあるように思えるかもしれませんが、DNAメチル化と同様に、それぞれの細胞や特定の遺伝子における適切なクロマチン環境の維持や変化を担う制御メカニズムが存在します。この役割を担う特別なタンパク質は、ヒストンの「リーダー」、「ライター」、「イレーサー」と呼ばれています。
ヒストン修飾のリーダー
ヒストンPTMのリーダータンパク質は、ヒストンテールの修飾部位に結合し、その情報を処理する酵素群です。このようなタンパク質の多くは、アセチル化アミノ酸残基に結合するブロモドメイン(bromodomain)、または、メチル化アミノ酸残基に結合するクロモドメイン(chromodomain)のいずれかを含んでいます。
リーダータンパク質が結合すると、クロマチンリモデリング複合体のような他の因子をリクルートすることにより局所的なクロマチン環境を変化させます。ブロモドメインを含むリーダーの一例としては、SWI/SNF (SWItch/Sucrose Non-Fermentable)複合体が知られています。SWI/SNFは、ヒストン-DNA相互作用を不安定化させることにより、DNA上のヌクレオソームの位置を変えることができるため「アクセスリモデラー」と呼ばれ、転写因子が結合しやすいように結合部位を露出させることにより、遺伝子発現を促進します。なお、SWI/SNFのサブユニットはがん細胞株ではしばしば欠損していることが確認されています。
クロモドメインを含むリーダーの例としては、CDH4 (Chromodomain-Helicase-DNA-binding protein 4)があります。CDH4は脱アセチル化酵素とヌクレオソームリモデリング複合体の一部であり、転写を停止させます。皮膚筋炎として知られる自己免疫疾患は、このタンパク質に対する自己抗体によって引き起こされます。
ヒストン修飾のライター
ヒストンPTMのライタータンパク質は、コアヒストン複合体に含まれるヒストンテール上のアミノ酸残基を積極的に修飾するクロマチン修飾酵素群です。これらの酵素には、ヒストンアセチルトランスフェラーゼ(HAT)、ヒストンメチルトランスフェラーゼ(HMT)およびリン酸化酵素が挙げられます。
HATファミリーの重要なメンバーのひとつが、細胞の増殖と分化を制御するp300です。p300酵素は4つのコアヒストンすべてをアセチル化することができ、クロマチンリモデリングを介して転写を制御します。さらに、転写因子と結合することにより、転写コアクチベーターとしても機能します。p300酵素は腫瘍の成長を防ぐために重要なことから、生物医学研究の興味深いターゲットになっています。
一方で、HMTファミリーの重要なメンバーは、エピジェネティックな転写抑制のメディエーターとしての機能を持つ EZH2 (enhancer of zeste homolog 2)です。ヒストンH3K27のモノメチル化、ジメチル化、トリメチル化を触媒し、ヘテロクロマチン形成と遺伝子機能のサイレンシングを誘導します。 FDAが最近承認したEpizyme Therapeutics社のタズベリク(Tazverik)と呼ばれるEZH2阻害剤は、特定のがん治療薬です。
ヒストン修飾のイレーサー
ヒストンPTMを消去するイレーサータンパク質は、それぞれのアミノ酸残基から修飾を除去します。これらの酵素には、ヒストン脱アセチル化酵素(HDACs)とヒストン脱メチル化酵素(HDMs)が含まれます。
ヒストン脱アセチル化酵素は、ヒストンテールからアセチル基を除去します。すると、アセチル化により電気的に中和されていたヒストンは再び正電荷を帯びるため、ヒストンとゲノムDNAの結合が促進されます(訳注:塩基性アミノ酸のリジンに富むヒストンテールは生理的pHにおいて正に帯電しています)。これにより、DNAの構造が凝縮し、転写が阻害されます。HDACファミリーは、それぞれの機能、およびDNA配列の類似性に基づいて5つのクラスに分類されています。“古典的”なHDACとして知られるクラスI、II、IVのメンバーは、トリコスタチンA (TSA, trichostatin A)により阻害される亜鉛イオン依存性の活性部位を持ちます。クラスIIIの酵素はTSAの影響を受けず、すべてNAD+ (nicotinamide adenine dinucleotide)依存性のタンパク質です。HDACの阻害剤は、気分安定薬や抗てんかん薬として精神医学の分野において長年使用されており、最も顕著な例はバルプロ酸(valproic acid)が挙げられます。
ヒストン脱メチル化酵素は、ヒストンテールからメチル基を除去します。長年、ヒストンのメチル化は不可逆と信じられていましたが、2004年にヒストン脱メチル化酵素LSD1が発見されると、次々に新たなヒストン脱メチル化酵素が発見されました。これらは大別して2つのサブファミリーに分けられます:KDM1A (K demethylase 1A)の属するFAD (flavin adenine dinucleotide)依存性のグループ と、JmjC型グループです(訳注:KDM1AはLSD1 (lysine specific demethylase 1)とも呼ばれます。JmjC型は、Jumonji C-terminal domain-containing histone demethylase: 略してJHDMファミリーともよばれます。名前に含まれるJumonjiはマウスの神経系の発生に関与する因子として同定されたJumonji 遺伝子(十文字)に由来します) 。モノメチル化あるいはジメチル化修飾を除去を触媒する酵素の他、トリメチル化された修飾の除去を触媒する酵素もあります。 転写に関与する他の多くのエピジェネティックタンパク質と同様に、ヒストン脱メチル化酵素の発現が変化すると、異常なヒストン修飾が生じ、がんの進行、転移、治療抵抗性などを引き起こす可能性が生じます。
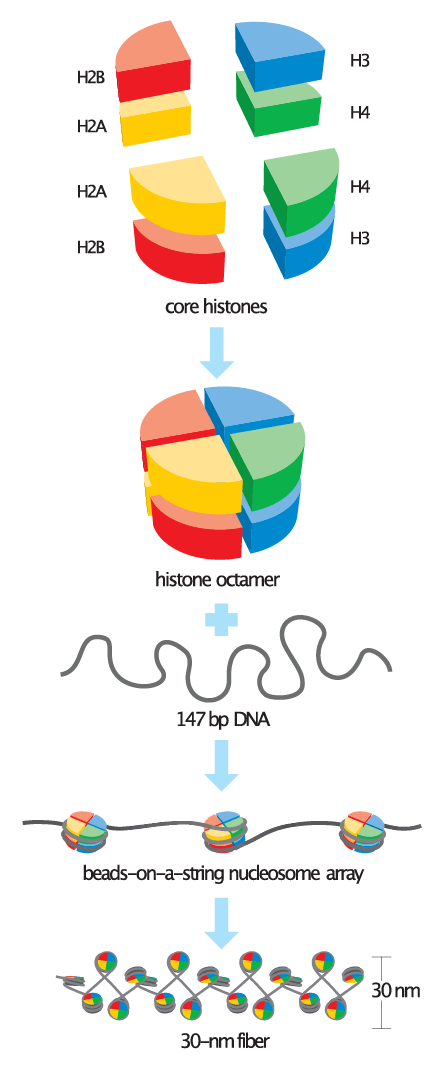
Image credit: Wikimedia Commons - Basic_units_of_chromatin_structure.svg - CC BY-SA 4.0
RNAのメチル化とその他の修飾
DNAのメチル化やヒストンPTMに加え、最近ではRNAにもエピジェネティクスに関与する転写後の化学修飾があることが明らかになっています。DNAからリボソームへ遺伝情報を伝えるメッセンジャーRNA (mRNA)、およびタンパク質に翻訳されない長鎖非コードRNA(lncRNA, long non-coding RNA)は、アデニンのN6位がメチル化されたm6A修飾を受ける可能性があります。
真核生物のmRNAにm6A修飾が起こるという初めての報告は1970年代にありましたが、2011年にGuifan Jiaらは、脂肪量と肥満に関連するタンパク質FTO (fat mass- and obesity-associated)をノックダウンするとmRNA中のm6A量が増加し、FTOを過剰発現させるとm6A量が減少することを発見し、一躍脚光を浴びました。
m6A修飾は真核生物で最も多く見られるRNA修飾の一つで、コンセンサス配列RR(m6A)CHで起こります(訳注:ここでRはアデニンまたはグアニンを、Hはグアニン以外の塩基を意味します)。この塩基修飾は、哺乳類では全てのmRNAにおいて平均〜3個存在し、細胞分化や個体の発生、様々な生物学的シグナル伝達やストレス応答に重要であることが示されています。この修飾は、mRNAの5’UTR、大きなエクソン、および3’UTRの終止コドン近傍に濃縮されています。
DNAメチル化やヒストンPTMの分野と同様に、RNA修飾にもリーダー、ライター、イレーサーが存在します。
RNAメチル化のリーダータンパク質は2つのファミリーに分類されます。YTHファミリー(YT521-B homology family)のようにm6Aを直接読み取るものもあれば、いくつかのhnRNP (heterogeneous nuclear ribonucleoprotein)のようにマークそのものではなく、RNA修飾により変化した二次構造を読み取るものも存在します。 YTHファミリーのうち、YTHDF2 (YTH domain family 2)の機能解析では、メチル化されたmRNAの細胞質局在の調節や分解に関与することが示されています。一方、YTHDF1は、翻訳開始を助けることによりメチル化されたmRNAの翻訳を促進します。他のリーダータンパク質は、メチル化されたmRNAの貯蔵や輸送、細胞内局在に影響を与えます。
最初のm6Aのライタータンパク質は1994年に発見されました。哺乳類細胞において非常に重要な2つライターは、METTL3 (methyltransferase-like 3)とMETTL14 (methyltransferase-like 14)です。これら2つのタンパク質は、METTL3-METTL14からなる安定なヘテロ二量体のコア複合体を形成し、核内のRNAに対するm6A修飾を担っています。METTL3がm6Aメチル化の触媒活性を担い、METTL14は基質認識に重要であり、複合体構造の安定化も担っています。
上述したFTOタンパク質の特徴の解明により、RNAのm6A修飾への関心が再燃しました。イレーサータンパク質であるFTOは、RNA分子のm6Aを脱メチル化しますが、この遺伝子の変化は、体重の増加と関連することが報告されています。もう一つのRNA脱メチル化酵素はALKBH5であり、FTOと同様にin vitroおよびin vivoにおいてmRNA中のm6A修飾を酸化的に解消します。Alkbh5 (alpha-ketoglutarate-dependent dioxygenase AlkB homolog 5)をノックアウトすると、雄マウスのmRNAにおけるm6Aレベルが上昇し、精子形成と生殖能力が損なわれることが報告されています。
m6A以外にも、N1-メチルアデノシン(m1A)、5-メチルシトシン(m5C)、シュードウリジン、2’-O-メチル化(2’OMe)など、哺乳類のmRNAには多くの修飾が存在しています。この化学修飾の組み合わせにより、RNA代謝と関連する生理学的プロセスのほぼすべての側面が調節され、真核生物、特に哺乳類における複雑な遺伝子発現の調節経路に新たな一面を加えています。
非コードRNAとエピジェネティクス
分子生物学のセントラルドグマに反して、タンパク質に翻訳されないRNAの一群が存在します。ncRNA (non-coding RNA: 非コードRNA)と呼ばれるこれらのRNAには、tRNA (transfer RNA: 転移RNA)やrRNA (ribosomal RNA:リボソームRNA)のほか、miRNAやsiRNA、piRNA、snoRNA、snRNA (small nuclear RNA: 核内低分子RNA)、exRNA (extracellular RNA: 細胞外RNA)、scaRNA (small Cajal body-specific RNA: 低分子カハール体特異的RNA)、lncRNAなどの様々な低分子RNAが含まれます。 miRNA、siRNA、piRNA、snoRNA、lncRNAなど、これらの多くがエピジェネティックな制御に関与しています(訳注:日本語名が併記されていないものは後述)。1980年代までは、ncRNAはおろか、RNA全般の制御作用についてはほとんど知られていませんでした。それが2000年代初頭にmiRNAが発見されると、ncRNA革命は勢いを増し、さらに多くのncRNAファミリーが発見されました。
マイクロRNA(microRNA, miRNA)
miRNA は、22ヌクレオチド長の一本鎖RNAです。標的となるmRNAと塩基対を形成することにより、転写後のRNAサイレンシング経路として作用し、転写産物の分解や翻訳の抑制に寄与します。miRNAは、primary miRNA (pri-miRNA)分子を起源とする約70-80ヌクレオチド長のRNA転写産物で、通常は標的遺伝子のイントロン領域に由来します。pri-miRNAはそれ自体が折り返って短いヘアピンを形成し、DICER (刻むものという意味)と呼ばれる酵素により切断されて成熟したmiRNAとなります。成熟したmiRNAは、RISC (RNA-induced Silencing complex: RNA誘導サイレンシング複合体)として知られるタンパク質複合体に取り込まれます。miRNAは相補的な配列をもつmRNAを選別する役割を担い、相同性の高いmRNAはRISC複合体により分解されます。
低分子干渉RNA (small interfering RNA, siRNA)
siRNAは20~27塩基対の小さな二本鎖RNA (dsRNA)であり、これらもRISCへと取り込まれて標的となるmRNAの選別機能を担います。siRNAは、DICER酵素により長いdsRNAが切断されることにより生成されます。siRNAがRISCに取り込まれると、その二本鎖はほどけて一本鎖RNAになります。このとき、熱力学的安定性により選択された一方の鎖(ガイド鎖)はRISC内部に残り、他方の鎖(パッセンジャー鎖)はRISCにより分解されます。こうしてガイド鎖を保持するRISCは、相補的な配列を持つmRNAを標的として結合し、それを分解します。
miRNAとsiRNAは、 mRNAとの相互作用においてやや異なる特性を示します。miRNAは標的mRNAと完全に相補的である必要はなく、RISC内でmRNAの分解や翻訳の抑制を引き起こすことが可能です。このRISCとmRNAの結合には、シード配列(約6塩基)と呼ばれる短い配列が一致していれば十分です。一方、siRNAは標的mRNAと完全に相補的でなければ機能しません。この違いにより、miRNAは多くの異なるmRNA (100種類以上)に結合し、それらの発現を調節することが可能です。一方、siRNAは特定のmRNAに対して完全に一致するため、通常は1種類のmRNAの発現調節のみに影響を与えます。
PIWI相互作用RNA (PIWI-interacting RNA, piRNA)
その他の非コードRNA分子についてはあまり知られていません。piRNAは21-31ヌクレオチド長の一本鎖低分子RNAであり、RISCとの複合体を形成することは知られていますが、その生成メカニズムはまだ完全には解明されていません。piRNAは哺乳類の精巣や卵巣に存在し、精子や胚の形成に関与していると考えられています。
核小体低分子RNA(small nucleolar RNA, snoRNA)
snoRNAは核小体に局在しています。前述のncRNA群とは異なり、その長さは60~300ヌクレオチドと範囲が広く、一定のサイズ幅を有していません。その主な機能は、mRNA、tRNA、rRNAのような他のRNA種に対する化学的RNA修飾を誘導することです。snoRNAは、Box C/D型とBox H/ACA型の2つのクラスに分けられ、それぞれ異なるRNA修飾を担います。 Box C/D型のsnoRNAは、塩基対A-UまたはC-Gからなるステム構造と特徴的な塩基配列box Cおよびbox Dを有する構造を形成し、メチル化に関連しています。一方、 Box H/ACA型のsnoRNAは、 2つのヘアピンを連結する特徴的な配列box Hと、3’側にACAの塩基配列を含むテールからなり、シュードウリジン化に関連しています。
長鎖非コードRNA (long non-coding RNA, lncRNA)
lncRNAは、200ヌクレオチド以上の長さを持ち、タンパク質に翻訳されないRNA転写産物として定義されています。lncRNAはエピジェネティックな遺伝子発現制御に関与しており、その代表的な例がX染色体不活性化です。哺乳動物のメスの発生初期において、2本あるX染色体の一方は、不活性化型のクロマチン修飾が多層的に付加されて沈黙状態となります。このプロセスはXist と呼ばれるlncRNAの発現から始まり、これによりH3K9のアセチル化の消失し、つづいてH3K27のメチル化が不可逆的に蓄積します。この結果、X染色体の一方を転写的に不活性化します。
エピジェネティクス研究の応用
近年、がんや糖尿病、肥満、さらには老化といった疾患におけるエピジェネティクスの重要性が次々に解明されてきました。DNAメチル化、転写後のヒストン修飾、および非コードRNAの変化は、さまざまな病態との関連が報告されています。
特に、DNA修復遺伝子や、がん抑制遺伝子のプロモーター領域に存在するCpGアイランドの過剰なメチル化は、それらの遺伝子の発現低下を引き起こすことが知られています。このような過剰メチル化は、膀胱がんや胃がん、甲状腺がん、大腸がん、脳腫瘍、肺がん、前立腺がん、乳がんなど、多くのがんで高頻度に確認されています。
また、ヒストンPTMやそれらの制御因子の異常も、腫瘍形成に関与しています。がん細胞では、これらの修飾が乱れることにより、がん遺伝子の異常な活性化や、逆に、がん抑制因子の異常な不活性化が引き起こされる可能性があります。
さらに、miRNAやlncRNAも、がんや心血管疾患、神経変性疾患の病態生理に関与していることが示されてきています。こうした知見を背景に、エピジェネティックな変化を診断や治療に応用しようとする研究が急速に進展しており、今後の疾患制御における新たなアプローチとして大きな注目を集めています。
エピジェネティクスに関する臨床研究の分野において、近年、おそらく最も関心を集めている領域の一つが、リキッドバイオプシー(液体生検)の解析です。リキッドバイオプシーは、患者に対し可能な限り非侵襲的に生体試料を採取する方法であり、主に血液や血漿、血清などが利用されますが、他の体液(尿や唾液など)が用いる場合もあります。リキッドバイオプシーは様々な種類のバイオマーカーを解析することができ、病気の進行や治療に対する反応をモニターしたり、病気の早期警告サインを探すための診断や予後検査に使用したりすることが可能です。
特に、リキッドバイオプシーは、がんの検出とモニタリングに非常に有用です。腫瘍(がん組織)は、DNA断片や腫瘍細胞を周囲の血液や尿、唾液などに放出するため、それらを解析することによりがんの存在や性質を明らかにすることが可能です。実際、リキッドバイオプシーでは、循環腫瘍細胞(circular tumor cells; CTC)や、がんに関連した変化を有する循環セルフリーDNA (cell-free DNA; cfDNA)の存在可能性が解析対象となります。当初のリキッドバイオプシーの解析は、主にがんに関連した遺伝子変異の検出を目的としていましたが、近年ではそれに加えてDNAメチル化パターンの変化、ヒストンやヌクレオソームの修飾状態、あるいはmiRNAの発現プロファイルといったエピジェネティックな変化を調べる方法としても大きな関心が寄せられています。
まとめ
エピジェネティクスは、遺伝子発現の制御、DNAの複製と修復、核内における高次の組織化など、多くの細胞機構に寄与する根源的な生物学的なプロセスです。そのメカニズムは、DNA配列やアミノ酸配列の変化ではなく、DNAやクロマチン関連タンパク質への修飾をともないます。「エピジェネティクス」という言葉やその背景にある考え方は20世紀初頭に誕生しましたが、この分野が最も注目されるようになったのは、近代的なDNAシーケンシング技術が導入され、ゲノムワイドなエピジェネティック因子の解析が可能になったからです。
エピジェネティクスとは、多層的に構成された遺伝子発現の制御ネットワークです。その第一層として、DNAのシトシン塩基の修飾(メチル化)があり、特に遺伝子プロモーター領域におけるメチル化は、遺伝子発現の抑制を引き起こします。このDNAメチル化は、さらにクロマチン修飾酵素群を誘導し、それらがヒストンテールに修飾を加えます。ヒストンテールは主にリジン残基において修飾されますが、これらの修飾は、「リーダー」、「ライター」、「イレーサー」と呼ばれる特化した酵素複合体によりダイナミックに制御されます。このようなヒストン修飾は、クロマチン領域の活性化または不活性化を引き起こし、最終的に遺伝子発現状態を長期にわたり規定します。
別の層として、mRNA内部に存在する6-メチルアデニン(m6A)修飾が挙げられます。m6Aは、mRNA分子の半減期(安定性)を制御し、ひいてはそのタンパク質の産生量の調整に寄与します。さらに、非コードRNA (miRNA, siRNA, piRNA, snoRNA, lncRNA)も遺伝子発現の調節因子として働きます。miRNAとsiRNAは、いずれも標的mRNAと相補的に結合することにより、mRNAの分解を誘導し、遺伝子発現を抑制する転写後RNAサイレンシング経路として機能します。一方、lncRNAはX染色体不活性化の過程において機能し、哺乳類のメスでは2本のX染色体のうち1本を恒常的に沈黙させる役割を果たしています。
これらのエピジェネティック因子は、生物の正常な発生過程や恒常性を維持するために極めて重要であり、また、環境変化を感知し、それに適応するための手段として機能しています。さらに、こうしたエピジェネティックな修飾は、次世代に受け継がれる可能性があることも明らかになってきています。
これまでに行われた多数の機能解析研究、およびエピジェネティック・マークやクロマチン修飾因子のゲノムワイドなマッピングにより、心血管疾患、発達障害、がんなど、さまざまなヒト疾患におけるエピゲノム機構の関与が明らかにされています。
これらのエピジェネティック因子のバランスが乱れると、生理機能に壊滅的な影響をもたらす可能性があります。現在、多くの研究が、リキッドバイオプシーを用いて異常なエピジェネティック因子を同定する手法の開発に取り組んでおり、疾患の早期診断や新たな治療介入の可能性が期待されています。
About the author

Stefan Dillinger, Ph.D.
Stefan was born in the Free State of Bavaria, Germany. After studying biochemistry in Ulm and Regensburg, he got his Ph.D. in the field of epigenetics, studying the distribution of heterochromatin around nucleoli during cellular senescence. As a graduate student he started his own German science podcast “The Random Scientist” and is now the host of Active Motif’s Epigenetics Podcast. When Stefan is not working at Active Motif or recording podcasts, he is a passionate runner (he finished the New York City Marathon in 3 hours 21 minutes!!) and loves to spend time with his wife and son.
Contact Stefan on LinkedIn with any questions, or to get running advice.
