Express RNA-Seq受託サービスの概要
Express RNA-Seqは、短期間で高品質なmRNA解析データを取得できる次世代シーケンス(NGS)サービスです。Poly(A)(Polyadenylated mRNA)を選択的に捉え、Tn5酵素を用いたタグメンテーションにより、効率よく正確な遺伝子発現解析を実現します。
ライブラリ作製では、mRNAをオリゴ-dTプライマーで逆転写し、得られたcDNAをTn5による酵素反応で断片化し、シーケンスアダプターを付加します (タグメンテーション)。この反応により、断片化とアダプター付加が同時に行えるため、データの質を保ちながら解析スピードを大幅に向上させることができます。
従来のRNA-Seq解析と比較すると短い納期で解析が可能となっており、スピードが求められる研究やスクリーニング、高スループット遺伝子発現解析に最適です。
Express RNA-Seq受託解析サービスの特長:
- スタンダード解析よりも短い納期(約4週間)
- mRNA解析に最適化 (Poly(A))
- バッチ間のばらつきを低減
- RNA抽出からデータ解析まで一貫したサービス
- 1500万~2000万ペアエンド (PE50)
- 論文掲載に利用可能な図表つき
RNA抽出とライブラリ調製からシーケンシング、データ解析に至るまで、一貫したトータルサポートを提供します。また、より深い生物学的知見を得るために、当社が誇る実績豊富なChIP-SeqやATAC-Seq解析と組み合わせることが可能です。これにより、特定のサンプルや実験モデルにおける遺伝子発現プログラムとその制御に関わるクロマチン構造を統合的に理解できます。
短い納期で高品質な解析を実現するExpress RNA-Seq
一貫した専門的サポート
エピジェネティクス研究の知見を活かし、トランスクリプトミクスやクロマチン解析の経験をもとに、高品質な遺伝子発現データの取得をサポートします。
信頼できる高品質データ
最適化されたライブラリ調製ワークフローと高性能シーケンスプラットフォームにより、一般的な遺伝子発現解析に十分対応可能な安定した性能を確保しており、少量サンプルからでも十分なデータ取得が可能です。
包括的な解析レポート
統計的に厳密な解析を行い、論文掲載に利用可能な図表や透明性の高いQC指標を含む解析レポートを提供します。
Express RNA-Seq受託解析サービスに含まれる内容:
- 細胞、組織からのtotal RNA抽出
- RNAの品質・完全性の評価 (RIN値による評価)
- オリゴ-dTプライマーを用いた逆転写
- Tn5酵素を用いたライブラリ作製とQC
- Illuminaプラットフォームによる次世代シーケンス
- 包括的バイオインフォマティクス解析
- 生データQC
- 参照ゲノムへのマッピング
- 遺伝子発現量の定量
- 差次的発現解析 (条件間比較解析)
- Gene Ontology解析 (GO解析)
- データ可視化 (PCAプロット、ヒートマップ、ボルケーノプロット
- マルチオミックス統合解析 (※統合解析費用が別途生じます)
- ChIP-Seq、ATAC-Seq、DNAメチル化解析など、追加のエピゲネティクス解析と統合可能
- 高度なバイオインフォマティクス解析により、クロマチン状態と転写制御の関係性などのメカニズム解析を支援
Express RNA-Seqは、短期間でのmRNA解析を実現しつつ、データの信頼性も確保します。助成金申請や論文投稿、スクリーニング研究など、スピーディーな結果が必要なプロジェクトに最適で、効率的なワークフローにより、4~5週間で有用なトランスクリプトームデータを取得することができます。
Express RNA-Seq Workflow
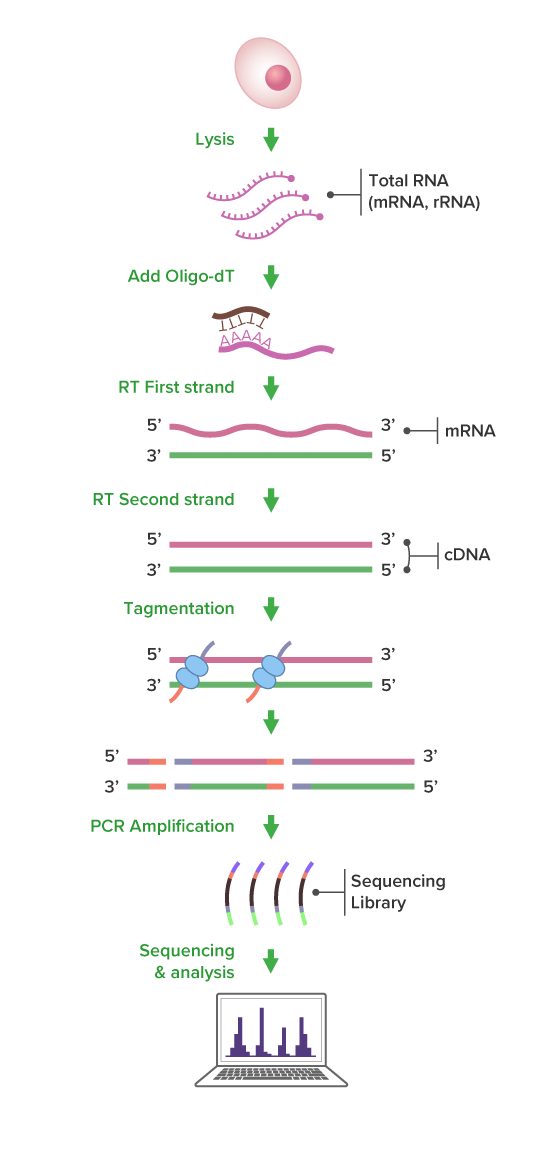
(Click image to enlarge)
Express RNA-Seqの解析データ
(Click image to enlarge)
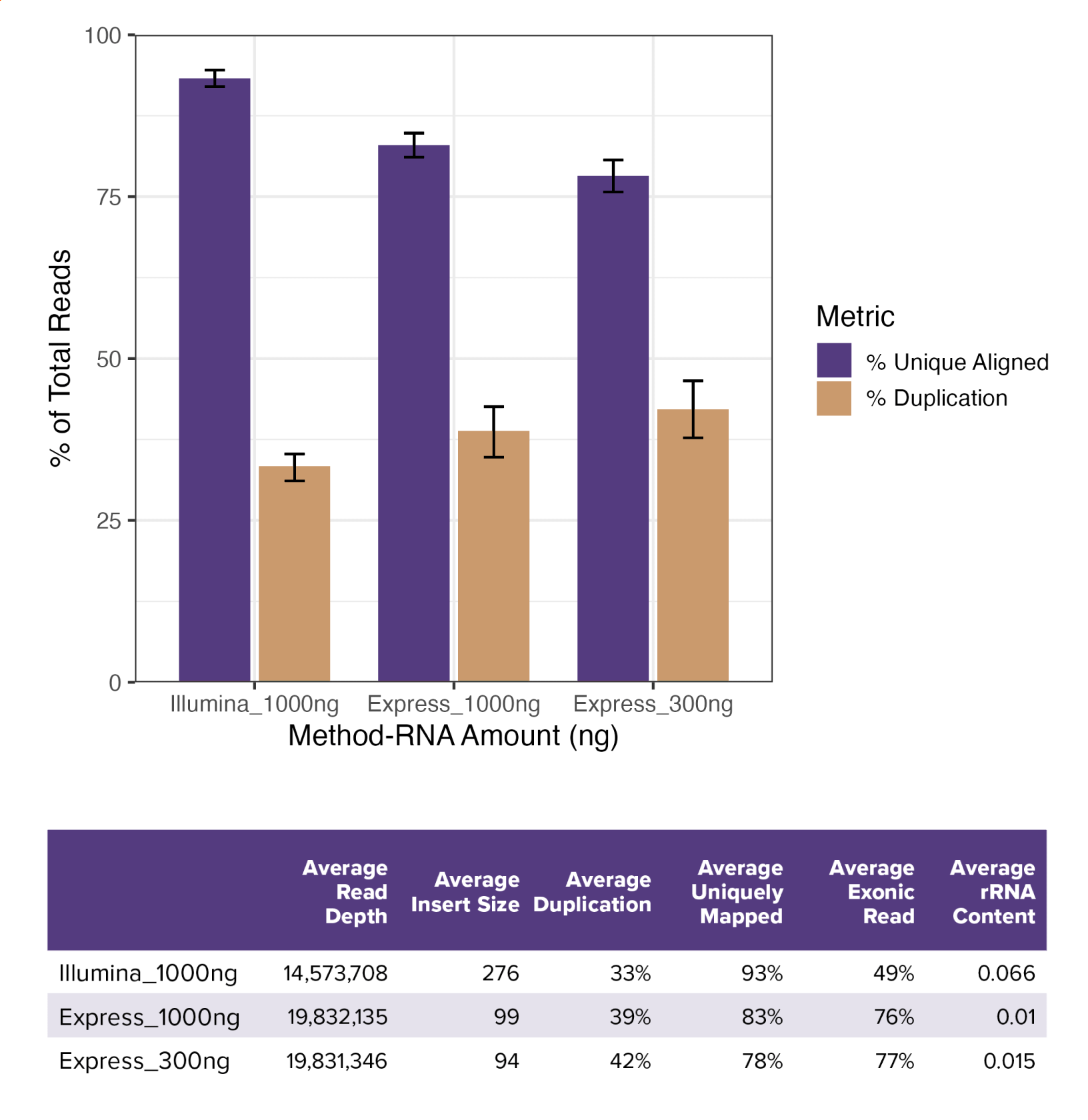
Fig. 1: Express RNA-SeqとIllumina Stranded mRNA Prepとの性能比較
LPS処理したTHP-1細胞から抽出したtotal RNAを用いて三重測定 (N=3) を行い、Express RNA-Seqでは300 ngおよび1,000 ng、Illumina Stranded mRNA Prepでは1,000 ngのtotal RNAを使用した。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqではPE50、Illumina Stranded mRNA PrepではPE150のペアエンドリードを使用して測定し、STARを用いて参照ゲノムにアラインメントした。Express RNA-Seqは、Illumina Stranded mRNA Prepに比べて一意にマッピングされるリード率がやや低く、重複率は少し高いものの、実用上十分な安定した性能を示した。シーケンス結果の概要は下表に示しており、Express RNA-Seqではエクソン領域にマッピングされるリードの割合が高く、mRNAをターゲットとしたライブラリ作製が効率的に行われていることが示された。
(Click images to enlarge)
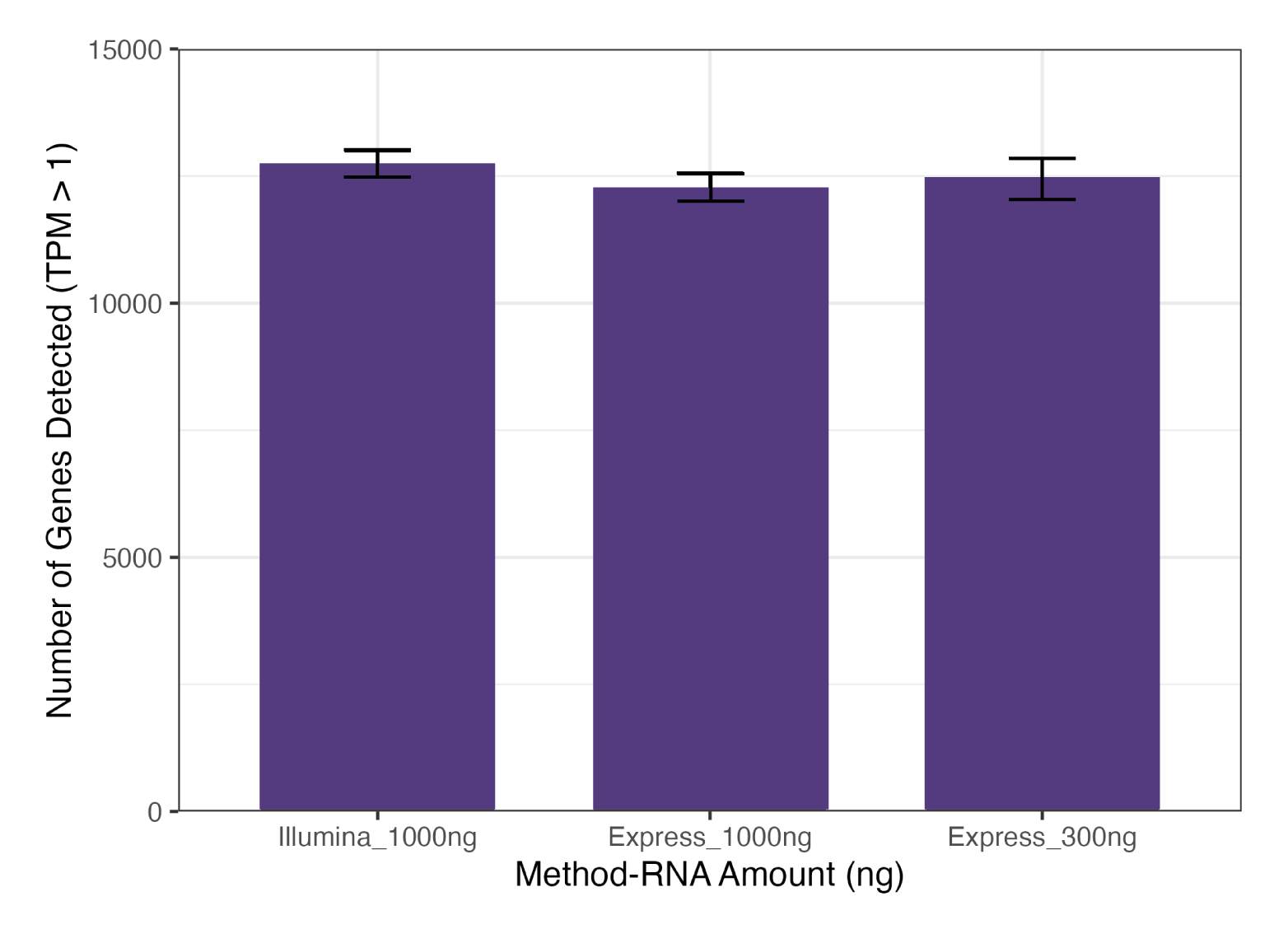
Fig. 2: Express RNA-SeqとIllumina Stranded mRNA Prepでの遺伝子検出数の比較
LPS処理したTHP-1細胞から抽出したtotal RNAを用いて二重測定 (N=2) を行い、Express RNA-Seqでは300 ngおよび1,000 ng、Illumina Stranded mRNA Prepでは1,000 ngのtotal RNAを使用した。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqはPE50、Illumina Stranded mRNA PrepはPE150のペアエンドリードを使用し、リードはSTARを用いて参照ゲノムにアラインメントした。結果、 Express RNA-Seqは、低いRNA量でもIllumina Stranded mRNA Prepとほぼ同等の遺伝子検出数を示した。
(Click image to enlarge)
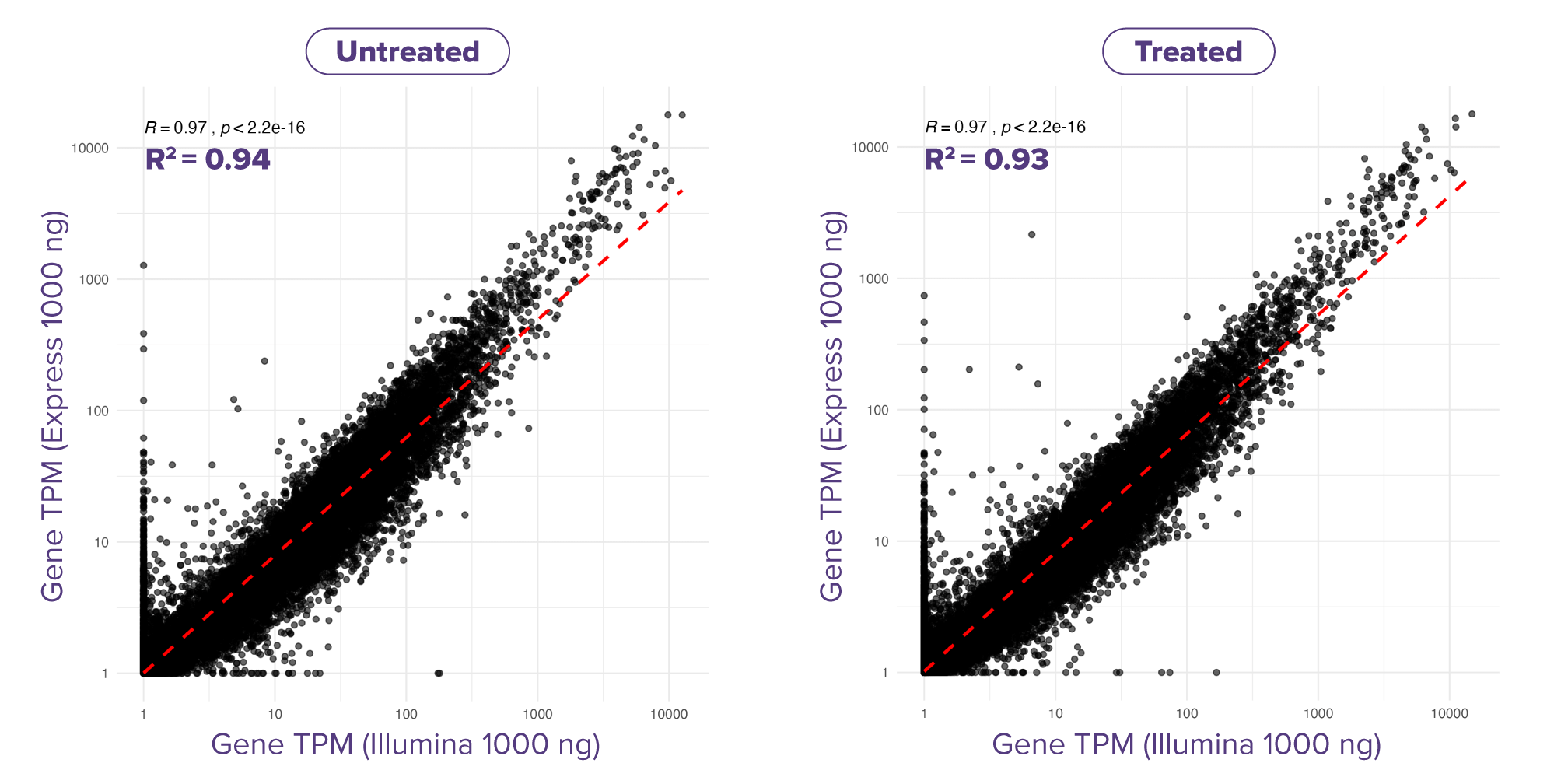
Fig. 3: Express RNA-SeqとIllumina Stranded mRNA Prepにおける遺伝子発現値の相関
LPS処理したTHP-1細胞から得たtotal RNA (各サンプル1,000 ng) を用い、二重測定 (N=2) で解析した。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqはPE50、Illumina Stranded mRNA PrepはPE150のペアエンドリードを使用し、リードはSTARを用いて参照ゲノムにアラインメントした。結果として、Express RNA-Seqで得られた遺伝子発現値は、Illumina Stranded mRNA Prepで得られた値と高い相関を示し、両手法間での高い相関が確認された。
 (Click image to enlarge)
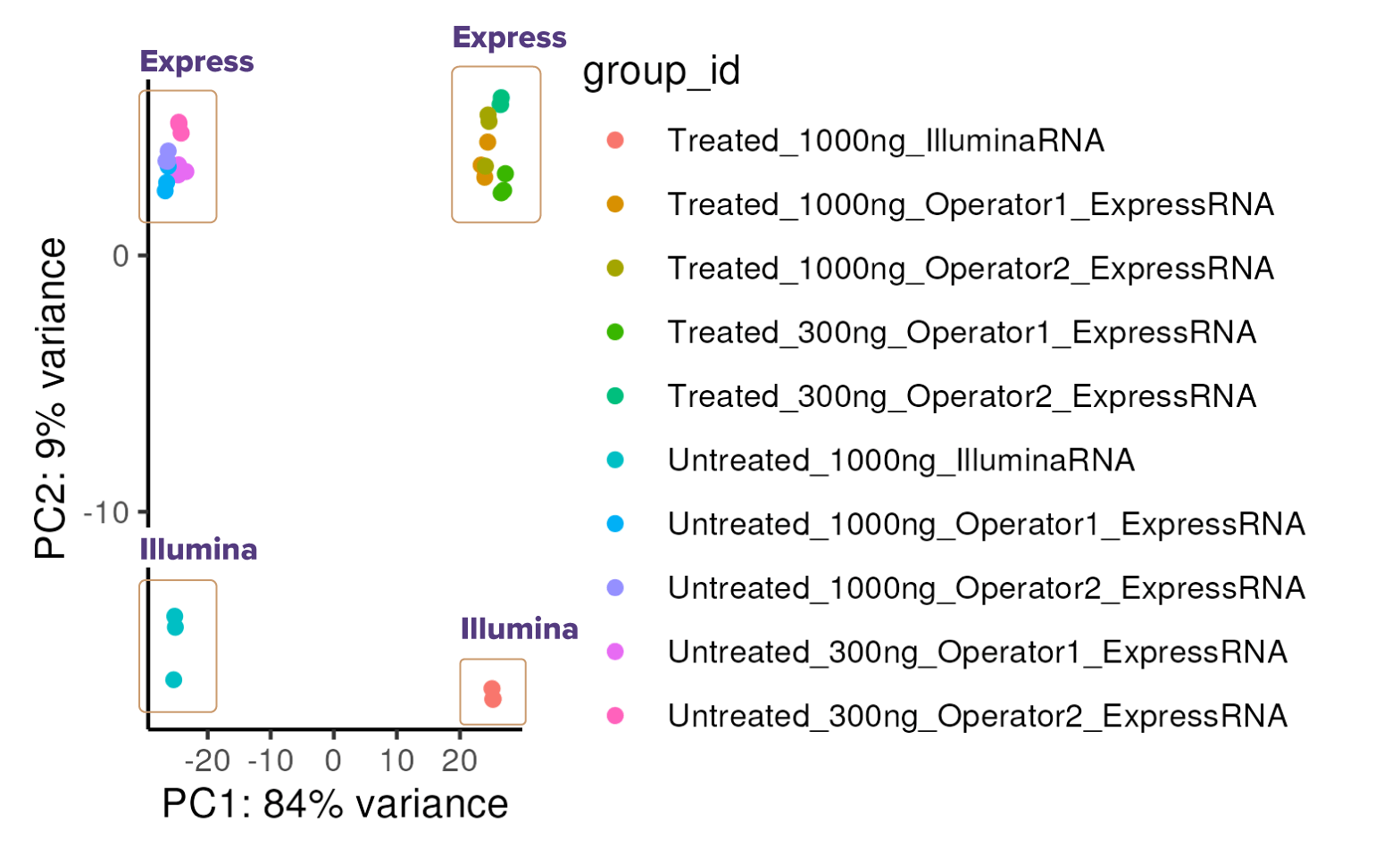
(Click image to enlarge)Fig. 4: Express RNA-SeqとIllumina Stranded mRNA Prepにおける遺伝子発現プロファイルの主成分分析 (PCA)
Express RNA-SeqおよびIllumina Stranded mRNA Prep-Seqで各サンプル1,000 ngのtotal RNAを用い、二重測定 (N=2) で解析した。さらに、Express RNA-Seqでは300 ngのtotal RNAを用いたサンプルも解析した。Express RNA-Seqの際2名のオペレーターがそれぞれ二重測定 (N=2) で解析している。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqはPE50、Illumina Stranded mRNA PrepはPE150のペアエンドリードを使用し、リードはSTARを用いて参照ゲノムにアラインメントした。結果、PCA解析により、未処理およびLPS処理したTHP-1細胞の遺伝子発現プロファイルを評価したところ、主要な変動要因はLPS処理であり (84%)、手法間の違いによる変動はわずか (9%) であることが確認できた。
(Click image to enlarge)
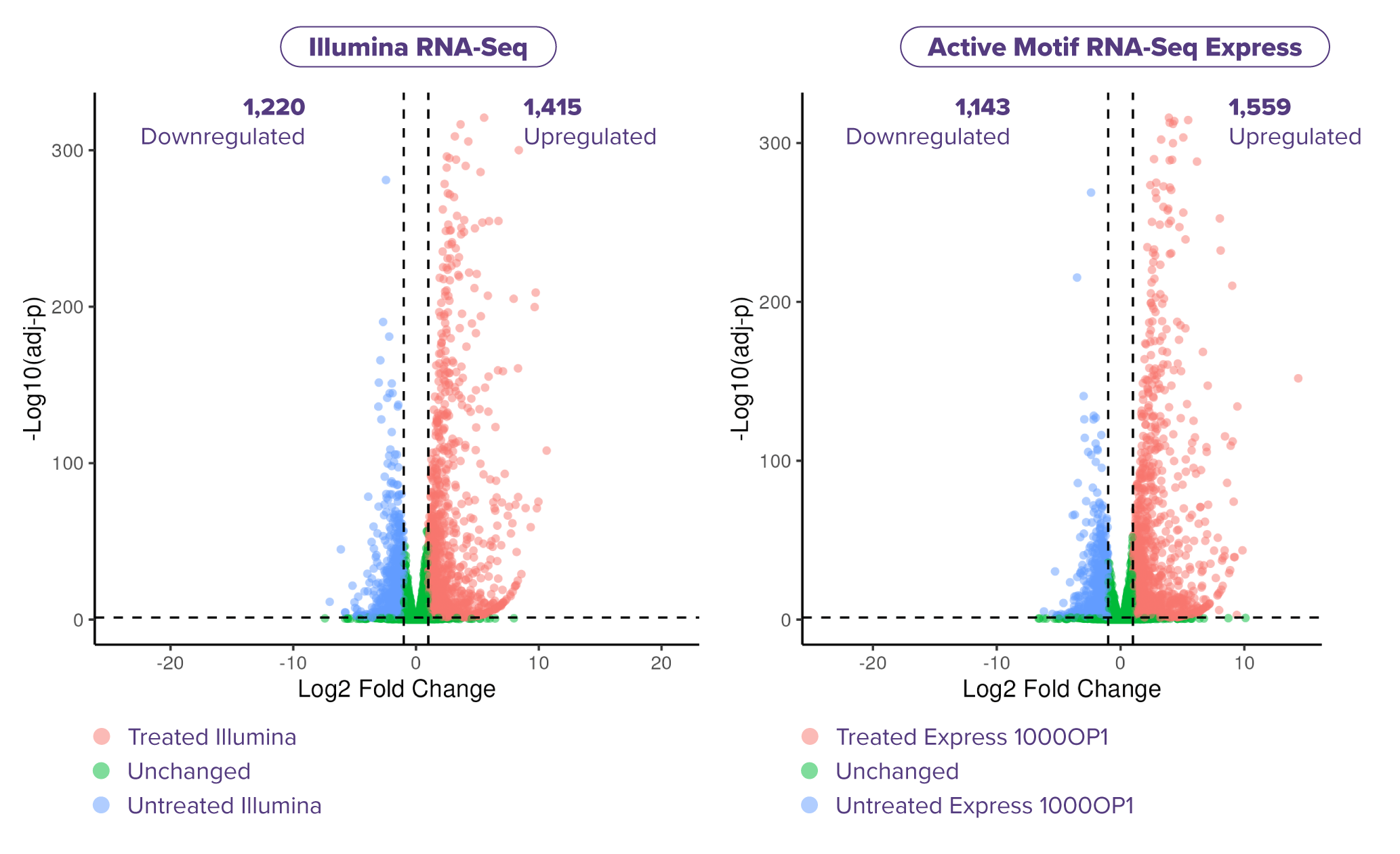
Fig. 5: Express RNA-SeqとIllumina Stranded mRNA Prepにおける差次的発現遺伝子の検出比較
LPS処理したTHP-1細胞から得たtotal RNA (各サンプル1,000 ng) を用い、二重測定 (N=2) で解析した。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqはPE50、Illumina Stranded mRNA PrepはPE150のペアエンドリードを使用し、リードはSTARを用いて参照ゲノムにアラインメントした。また、差次的発現遺伝子についてはDESeq2を用いて同定した。結果、LPS処理したTHP-1細胞における差次的発現遺伝子のボルケーノプロットを示したところ、Express RNA-SeqとIllumina Stranded mRNA Prepで検出される上方および下方に発現変動する遺伝子の数はほぼ同等であることが示された。
 (Click image to enlarge)
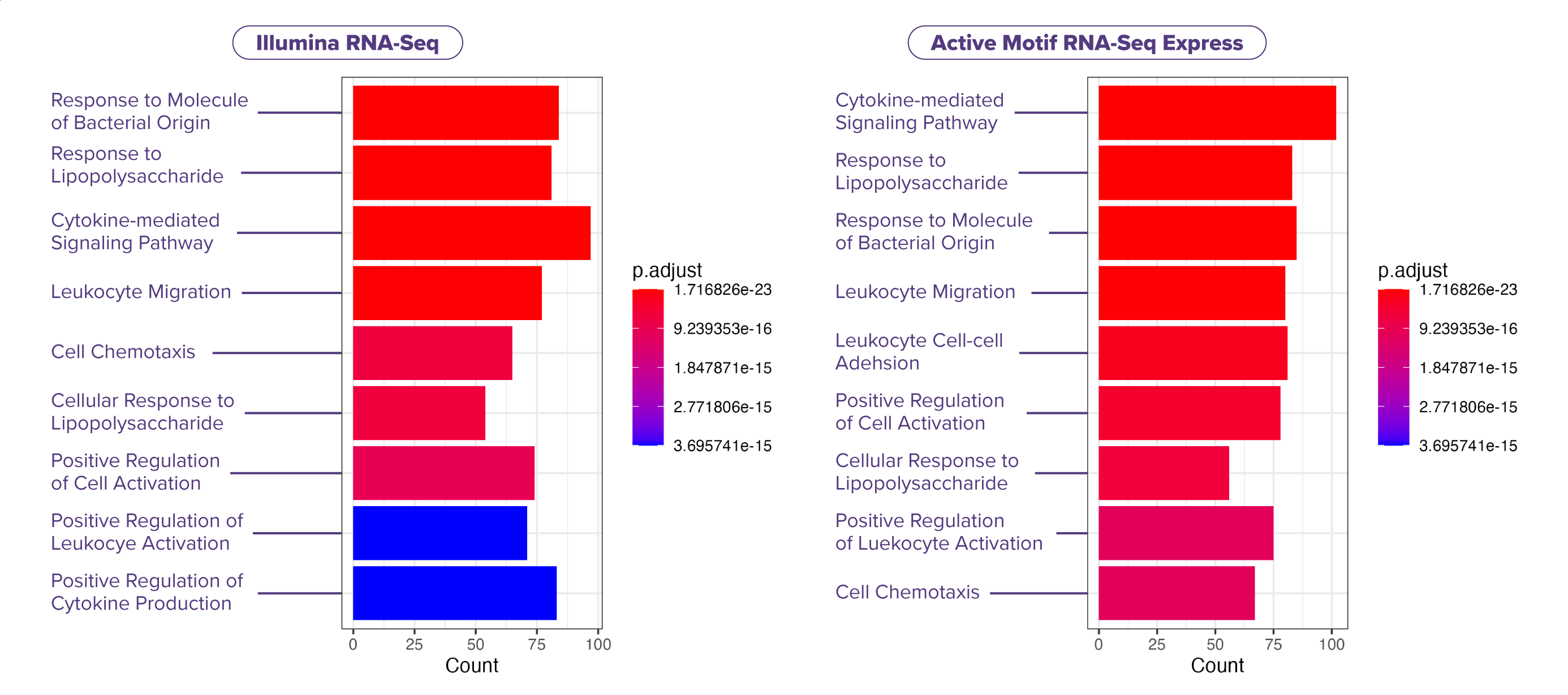
(Click image to enlarge)Fig. 6: Express RNA-SeqとIllumina Stranded mRNA Prep間における発現増加遺伝子のパスウェイ解析
LPS処理したTHP-1細胞から得たtotal RNA (各サンプル1,000 ng) を用い、二重測定 (N=2) で解析した。各遺伝子にマッピングされたリード数は約1,500万リードで、Express RNA-SeqはPE50、Illumina Stranded mRNA PrepはPE150のペアエンドリードを使用し、リードはSTARを用いて参照ゲノムにアラインメントした。また、差次的発現遺伝子はDESeq2を用いて同定した。LPS処理したTHP-1細胞におけるパスウェイ解析を実施したところ、Express RNA-SeqとIllumina Stranded mRNA Prepの両手法で、同様な発現上昇を示す生物学的プロセス (GOの分類) のサブセットが検出された。
Express RNA-Seq受託解析サービスのFAQ
1. Express RNA-Seq受託解析サービス全般
Express RNA-Seqは、迅速かつ高精度でスケーラブルな遺伝子発現解析を可能にする次世代シーケンス (NGS) ベースのRNAシーケンシング手法です。
本手法では、まずRNAを相補的DNA (cDNA) に変換し、その後タグメンテーションを用いたライブラリ調製を行います。このステップでは、Tn5酵素を用いてcDNAを同時に断片化しながら、同時にシーケンスアダプターを付加します。1回のタグメンテーション反応で行えるため、作業時間を短縮でき、手順も簡略化できることで、ワークフロー全体の効率が向上します。
Express RNA-Seqでは、以下の解析が可能です:
- 遺伝子発現量の定量
- 一部の選択的スプライシングイベントの検出
- 既知転写産物の同定
- 生物学的条件間での差次的遺伝子発現解析
Express RNA-Seqは、高スループットのトランスクリプトーム解析、バイオマーカー探索、比較遺伝子発現解析に適しています。
標準的なIlluminaベースのmRNA-Seqは、ポリアデニル化された転写産物を対象とした解析ワークフローであり、ゲノム全体をカバーすることで、既知および新規の転写アイソフォームやスプライスバリアント、アレル特異的発現の検出が可能です。そのため、包括的なトランスクリプトーム解析に適しています。
一方、Express RNA-Seqは、スピードと効率を重視して最適化された迅速なシーケンス手法です。タグメンテーションを用いたライブラリ調製により、cDNAの断片化とシーケンスアダプターの付加を同時に行うことで、作業時間を短縮します。さらに、PE50などの効率的なペアエンドリード構成を採用することで、短期間で高品質な遺伝子発現データを取得できます。
このように、IlluminaベースのmRNA-Seqが網羅性と深度を重視するのに対し、Express RNA-Seqは迅速性と効率性を重視した解析に適しています。
4~5週間です。具体的な納期については、サンプル輸送のタイミングによって変動することもありますが、約4週間となります。なお、ご提出いただいたサンプルの品質、希望するシーケンス深度などによって納期がかかる場合がございます。
2. 検体要件とRNAの品質について
Express RNA-Seqの検体要件は、精製RNAを提出するか、細胞ペレットまたは組織を提出するかによって異なります。
- 最小提出量:total RNA 500 ng / 哺乳類細胞 25,000~50,000個 / 組織 20 mg
- 推奨提出量:最大1 µgのtotal RNA /哺乳類細胞 50,000~100,000個 / 組織 50 mg
より多くのtotal RNAをご提供いただくことで、ライブラリの複雑度、転写産物の検出感度、およびデータ全体の信頼性が向上する傾向がありますので、表の範囲内で可能な限り多くご提出いただくことを推奨します。なお、ライブラリ調製前に、RIN値およびQubitを含むRNA品質管理 (QC) を実施します。
| サンプルタイプ | 推奨量 | 最小 | 多くご提出いただく理由 |
|---|---|---|---|
| Total RNA | 最大 1 µg | 500 ng | ライブラリの複雑性が向上し、転写産物の検出感度およびデータ全体の信頼性が高まります |
| 哺乳動物細胞 | 50,000~100,000細胞 | 25,000~50,000細胞 | シーケンスに適した十分なRNA量を確保できます |
| 凍結組織 | 50 mg以上 | 20 mg以上 | RNAの回収量を確保し、安定したライブラリ品質が得られます |
標準的なmRNA-Seqでは、RIN値7以上を推奨しています。
RIN値が低いサンプルについては、お客様のご了承をいただいた上で解析を行うことも可能です。
はい。凍結細胞ペレットおよび新鮮凍結組織からのRNA抽出が可能です。その他のサンプル種別については、こちらからお問い合わせください。
サンプルを「サンプル調製手順」に従って調製してください。サンプルを送付する際は、別途、ウェブでの解析申し込みやご提出いただく書類などがありますので、必ず日本法人 ([email protected]) までご連絡ください。
3. シーケンス深度と解析設定について
通常、1サンプルあたり1,500万〜2,000万リードのシーケンスを行います。このリード数は、遺伝子発現の正確な解析や差次的発現解析に十分な深さです。
ペアエンド50 bp(PE50)は標準的な構成であり、信頼性の高いカバレッジと効率を提供します。
より高解像度の用途には、ペアエンド150 bp(PE150)もオプションとしてご利用いただけます。
4. バイオインフォマティクス解析および納品物について
Express RNA-Seqのバイオインフォマティクス解析パイプラインには、通常以下の解析が含まれます:
- 生データの品質管理(FastQC):リードの品質を評価
- アダプターのトリミング:シーケンスアダプターを除去
- リファレンスゲノムへのアラインメント:リードを正確にマッピング
- 遺伝子・転写産物の定量:発現量を測定
- 差次的発現解析:有意な発現変動を示す遺伝子を特定
- Gene Ontology解析(GO解析):生物学的意義を解釈
- データ可視化:PCAプロット、ヒートマップ、ボルケーノプロット
この包括的な解析パイプラインにより、高品質の解析データ、有用な知見、下流解析や論文掲載に使用できる図表などを提供します。
Express RNA-Seq受託解析サービスで通常提供される納品物には、以下が含まれます:
- 生FASTQファイル:元のシーケンスリード
- アライン済みBAMファイル:リファレンスゲノムにマッピングされたリード
- カウントマトリクス:遺伝子発現量の定量
- 差次的発現テーブル:条件間の比較結果
- QCレポート:シーケンスおよびアラインメントの品質管理指標
- バイオインフォマティクス要約レポート:包括的な解析と結果の解釈
これらの納品物により、下流解析に必要なデータセットが揃います。生データと解析済みデータの両方を受け取ることができるため、さらなる研究や他のオミクス解析との統合にすぐ利用できます。
はい。Express RNA-Seqサービスには、論文掲載用の図も含まれています。
例えば:
- PCAプロット:サンプル間のクラスタリングや分散を可視化
- ボルケーノプロット:差次的発現遺伝子を強調
- ヒートマップ:サンプル間の発現パターンを表示
- パスウェイエンリッチメントプロット:関与する生物学的経路を要約
これらの図はすべて、論文掲載や下流解析に適した形式で作成されており、RNA-Seqの結果をすぐに提示・解釈できる状態で提供されます。
はい。Express RNA-Seqデータは、ChIP-Seq、ATAC-Seq、DNAメチル化データ、CRISPRスクリーニングなど、複数のオミクスデータセットと統合することが可能です。RNA-Seqを他のオミクスデータと統合することで、遺伝子発現制御や生物学的経路に関するメカニズムについて、より深い知見を得ることができます。
Express RNA-Seq受託解析サービスの資料
Express RNA-Seq Service Sample Submissionポータル
オンラインSample Submissionポータルは、解析したいサンプルの情報をアクティブ・モティフの担当者と共有し、依頼者には解析の進捗をご覧いただけるサイトです。 解析を依頼するために必要な連絡先、サンプルの情報、また解析の内容を記載していただきます。記載の方法や、サンプル提出方法について、ご不明な点がありましたら「[email protected]」までご連絡ください。
こちらもご参照ください:
| Name | Cat No. | 価格 (税抜) | |
|---|---|---|---|
| Express RNA-Seq | 25218 | Get Quote |