Single-Cell CUT&Tag Multiome Service Overview
Single-Cell CUT&Tag Multiome combines single-cell RNA expression profiling with CUT&Tag to simultaneously measure gene expression and histone modification localization in individual cells. CUT&Tag Multiome provides high-resolution insight into regulatory mechanisms governing gene expression.
The workflow begins with a bulk CUT&Tag reaction to label the chromatin feature of interest in isolated nuclei. Labeled nuclei are then encapsulated into individual 10x Genomics GEMs, where each nucleus is assigned a unique barcode during reverse transcription. This enables parallel capture of chromatin profiling and transcriptomes from the same cell. Independent single-cell CUT&Tag and scRNA-seq libraries are subsequently generated, sequenced, and jointly analyzed to provide an integrated view of epigenomic and transcriptomic states at single-cell resolution.
By directly linking chromatin states with gene expression in the same cell, Single-Cell CUT&Tag Multiome enables more precise interpretation of regulatory mechanisms driving cell identity and transcriptional programs.
Single-Cell CUT&Tag Multiome Highlights
- Simultaneous transcriptome and epigenome profiling. Measure gene expression and histone modification occupancy in the same single cell.
- High-quality single-cell data. Targeting ~5,000 cells with ~30,000 reads per cell for both the CUT&Tag and RNA libraries.
- Scientific and bioinformatic support. Guidance throughout the project, including data review and interpretation.
Active Motif’s Single-Cell CUT&Tag Multiome service includes:
- Nuclei preparation
- Bulk CUT&Tag reaction
- 10X Genomics GEM generation
- Reverse transcription and single cell barcode ligation
- Single-cell CUT&Tag and single-cell RNA-Seq library preparation
- Illumina Sequencing
- Comprehensive bioinformatic analysis and data delivery
When you partner with Active Motif, every project is supported by our team of scientists and bioinformatics specialists. We provide scientific guidance during experimental design, sample submission, and data generation to help ensure optimal project execution. After data delivery, our bioinformatics team is available to review your results, discuss quality metrics, and assist with data interpretation.
Single-Cell CUT&Tag Multiome Workflow

(Click image to enlarge)
Single-Cell CUT&Tag Multiome Service Data
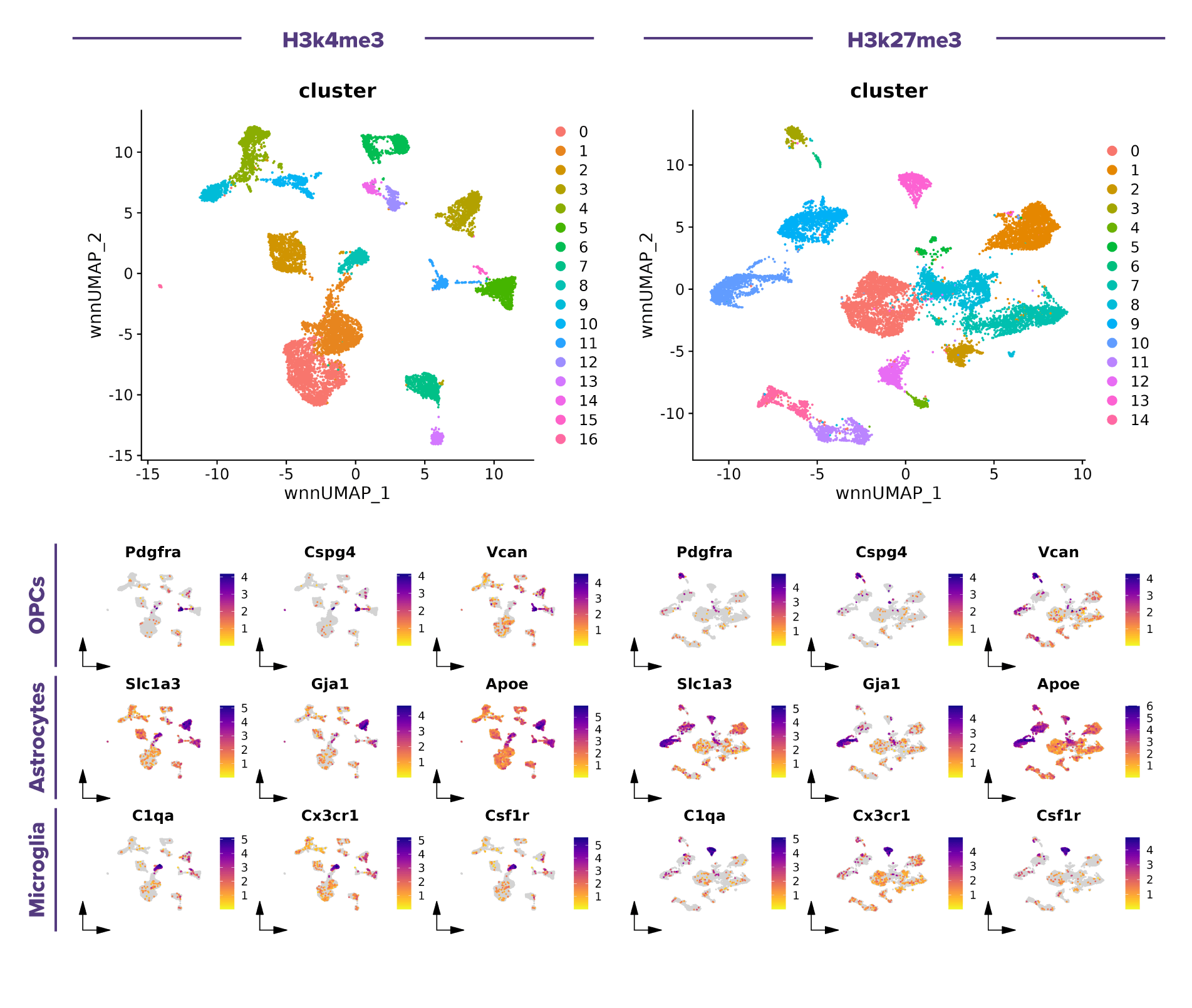
(Click image to enlarge)
Figure 1. Single-cell multiome (scRNA-seq + scCUT&Tag) analysis reveals cell type-specific histone modification landscapes in the mouse brain.
Top, weighted nearest neighbor (WNN) UMAPs showing cell clusters identified by integrating scRNA-seq and scCUT&Tag data for the H3K4me3 (left) and H3K27me3 (right) datasets. Bottom, scRNA-seq feature plots of representative cell type specific marker genes, including e.g., Pdgfra, Cspg4, Vcan (oligodendrocyte precursor cells, OPCs), Slc1a3, Gja1 and Apoe (astrocytes), and C1qa, Cx3cr1, and Csf1r (microglia). These marker genes exhibit distinct cell type-specific expression patterns, supporting the annotation of major glial cell populations and providing a transcriptional reference for interpreting the corresponding H3K4me3 and H3K27me3 chromatin landscapes.
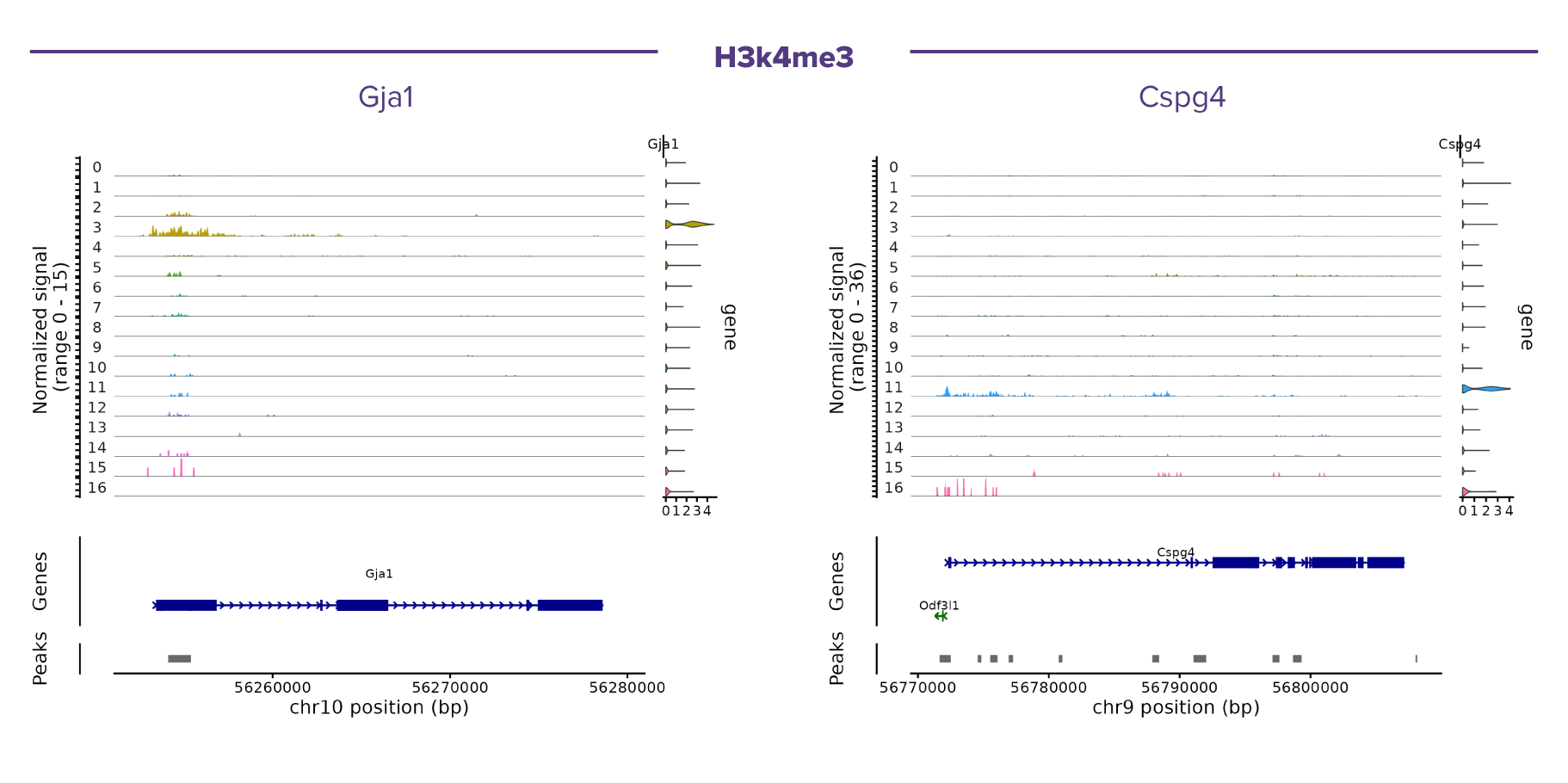
(Click images to enlarge)
Figure 2. Representative single-cell CUT&Tag coverage profiles of H3K4me3 at cell type-specific marker loci.
Pseudobulk coverage tracks are shown for each cell cluster, with clusters ordered as in the integrated UMAP. Normalized H3K4me3 signal is displayed above the corresponding gene annotation and identified CUT&Tag peaks. The examples illustrate cell type-specific enrichment of H3K4me3 at promoter regions of canonical marker genes, including Gja1 (astrocytes) and Cspg4 (oligodendrocyte precursor cells, OPCs). Consistent with the role of H3K4me3 as an active promoter-associated histone modification, strong enrichment is observed in the corresponding cell populations, supporting the concordance between chromatin state and cell identity inferred from the integrated scRNA-seq and scCUT&Tag analysis.
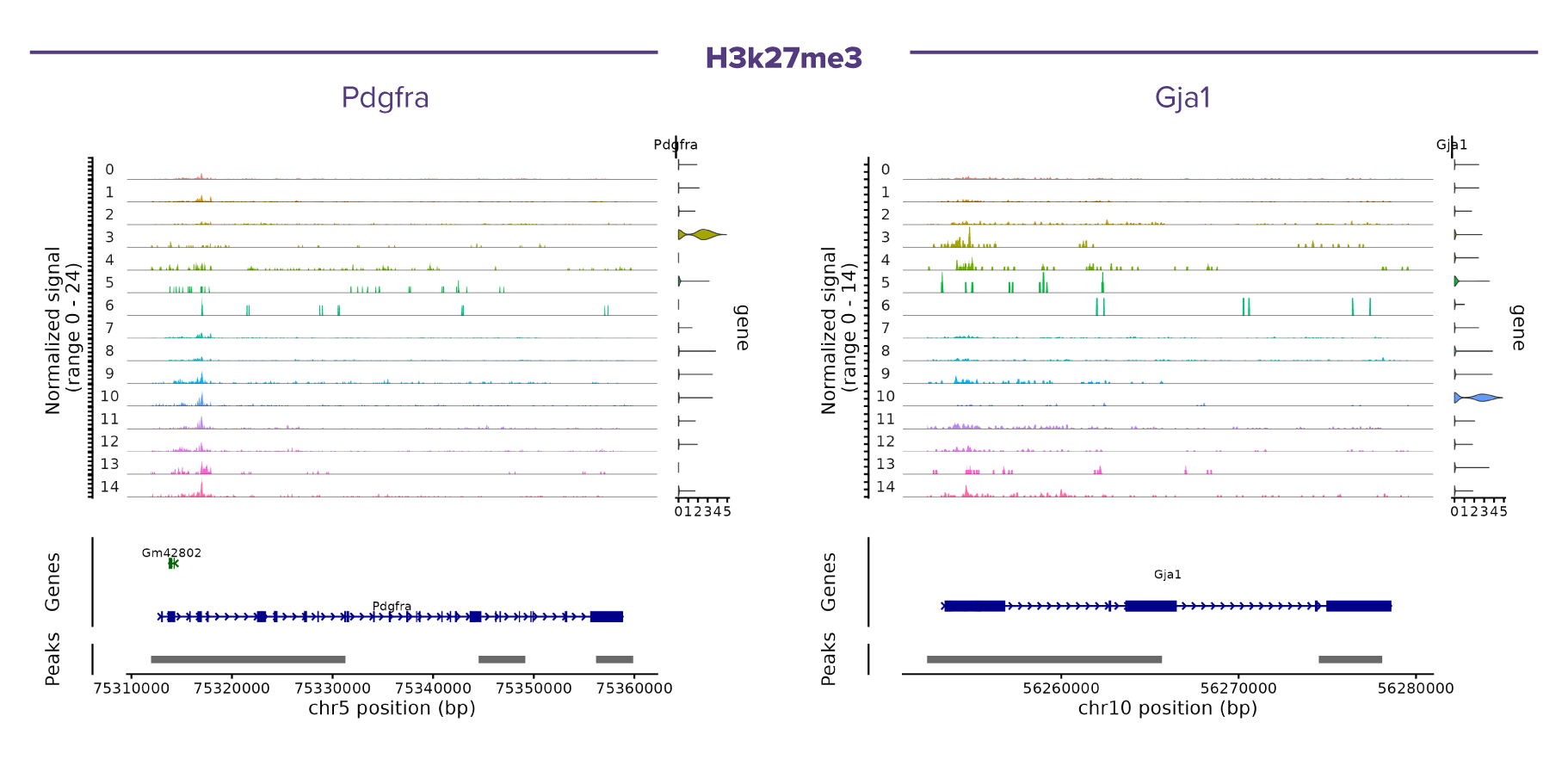
(Click image to enlarge)
Figure 3. Representative single-cell CUT&Tag coverage profiles of H3K27me3 at cell type-specific marker loci.
Pseudobulk coverage tracks are shown for each cell cluster, with clusters ordered as in the integrated UMAP. Normalized H3K27me3 signal is displayed above the corresponding gene annotation and identified CUT&Tag peaks. The examples highlight Pdgfra (oligodendrocyte precursor cells, OPCs) and Gja1 (astrocytes), illustrating the broad genomic distribution characteristic of the repressive histone mark H3K27me3. Cell type-specific differences in H3K27me3 occupancy are observed across clusters, reflecting distinct epigenetic states associated with transcriptional repression and cellular identity in the mouse brain.
Single-Cell CUT&Tag Multiome Service FAQs
1. General Overview
Single-Cell CUT&Tag Multiome is an advanced multiomics technology that simultaneously profiles histone modifications and gene expression in the same individual cell. By combining single-cell CUT&Tag epigenomic profiling with single-cell RNA sequencing (scRNA-seq), this approach directly links chromatin state to transcriptional activity, enabling researchers to identify cell types, characterize cellular heterogeneity, discover regulatory mechanisms, and study how epigenetic changes influence gene expression
Our Single-Cell CUT&Tag Multiome service has been validated for the following histone modifications:
- H3K27me3: A repressive histone mark associated with gene silencing and facultative heterochromatin.
- H3K4me3: An active promoter mark commonly enriched at transcription start sites (TSSs) of actively transcribed genes.
- H3K27ac: An active chromatin mark found at promoters and enhancers, associated with active gene expression.
Additional histone modifications may be evaluated upon request. Please contact us to discuss your specific research needs.
Yes. In addition to our validated targets (H3K27me3, H3K4me3, and H3K27ac), we offer custom antibody validation for researchers interested in profiling other histone modifications or chromatin-associated proteins.
Our scientists will evaluate your antibody for compatibility with the Single-Cell CUT&Tag Multiome workflow by assessing factors such as signal quality, specificity, background levels, and overall assay performance. If the antibody meets our quality criteria, it can be incorporated into your study.
Please contact our scientific support team to discuss your target, sample type, and project goals. We will work with you to determine the best validation strategy before initiating your study.
2. Sample Requirements
The Single-Cell CUT&Tag Multiome Service is compatible with a wide range of primary cells and tissue-derived samples, provided they can be prepared as a high-quality single-cell suspension. We have successfully validated the workflow using human PBMCs and mouse brain tissue, and many other primary cells and tissue types are also suitable depending on sample quality and dissociation efficiency.
For new or challenging tissue types, we also offer a Single-Cell Nuclei Preparation Test prior to full service processing. This optional step allows us to evaluate whether high-quality nuclei can be recovered from your sample, helping to de-risk the workflow before proceeding with the full Multiome assay.
If you're unsure about sample compatibility, our team is happy to review your project and provide recommendations.
For optimal results, we recommend submitting 250,000 to 2 million cryopreserved cells or 20–50 mg of frozen tissue. This ensures sufficient material for generating a high-quality single-cell suspension and robust multiomic data. If your sample falls outside these recommendations, our team can help determine the best approach for your project.
We load 7250 nuclei per sample in each lane to target 5000 nuclei in the data set which includes a multiplet rate of ~2%. Upon request, we can load more nuclei per sample. For example, targeting 10,000 nuclei for each sample requires us to load 14,500 nuclei per lane. However, this requires double the sequencing depth of a standard project and carries with it a ~4% multiplet rate.
The Single-Cell CUT&Tag Multiome Service simultaneously measures histone modifications and gene expression in the same individual cells, providing single-cell resolution to uncover cellular heterogeneity, identify rare cell populations, and directly link epigenetic regulation to transcription. In contrast, the Bulk CUT&Tag Service profiles histone modifications across an entire cell population, generating an average epigenetic landscape that is ideal for homogeneous samples and population-level comparisons.
3. Sequencing
We recommend a sequencing depth of approximately 25,000 reads per cell for both the CUT&Tag and gene expression (GEX) libraries. This level of coverage provides a balanced depth for robust detection of histone modification signals and transcriptomic profiles while maintaining efficient data quality and cost-effectiveness.
For the Single-Cell CUT&Tag Multiome Service, we recommend paired-end 2 × 50 bp sequencing for both libraries:
- RNA (gene expression) library: Paired-end 2 × 50 bp
- CUT&Tag (histone modification) library: Paired-end 2 × 50 bp
This sequencing configuration provides high-quality data for both gene expression profiling and histone modification mapping while supporting accurate cell identification and efficient downstream analysis.
With Active Motif’s end-to-end Single-Cell CUT&Tag Multiome Service, you receive a complete multiomic data package including raw sequencing data (FASTQ files), processed gene expression (GEX) and CUT&Tag count matrices, quality control (QC) metrics, and a comprehensive bioinformatics report. This integrated output enables direct downstream analysis of both epigenetic and transcriptional profiles at single-cell resolution.
Single-Cell CUT&Tag Multiome Service Documents
Single-Cell CUT&Tag Multiome Service Sample Submission Portal
Our online sample submission portal allows you to easily upload your service project samples and track your project status. Follow the sample submission instructions in the portal to ensure that all your samples arrive at Active Motif in the best possible condition and properly associated with your project.